DIRK BAKOWIES | HOME PAGE

Tel: +41 78 856 61 45 |
| Welcome to Dirk's homepage | ||||||
|
Research Interests |
|
|
|
Selected recent and past projects |
|
|
|
|
|
Water cavities | ||||
|
|
FABP | |||||
|
|
Carbopeptoids | |||||
|
|
Enzymes & Free Energies | |||||
|
|
||||||
|
|
|
Cavity analysis | ||||
|
|
Pair list algorithms | |||||
|
Experience |
|
|
CBS extrapolation | |||
|
|
ATOMIC | |||||
|
|
Error and uncertainty (1) | |||||
|
|
Error and uncertainty (2) | |||||
|
|
ATOMIC-2 | |||||
|
|
||||||
|
|
|
Giant Fullerenes | ||||
|
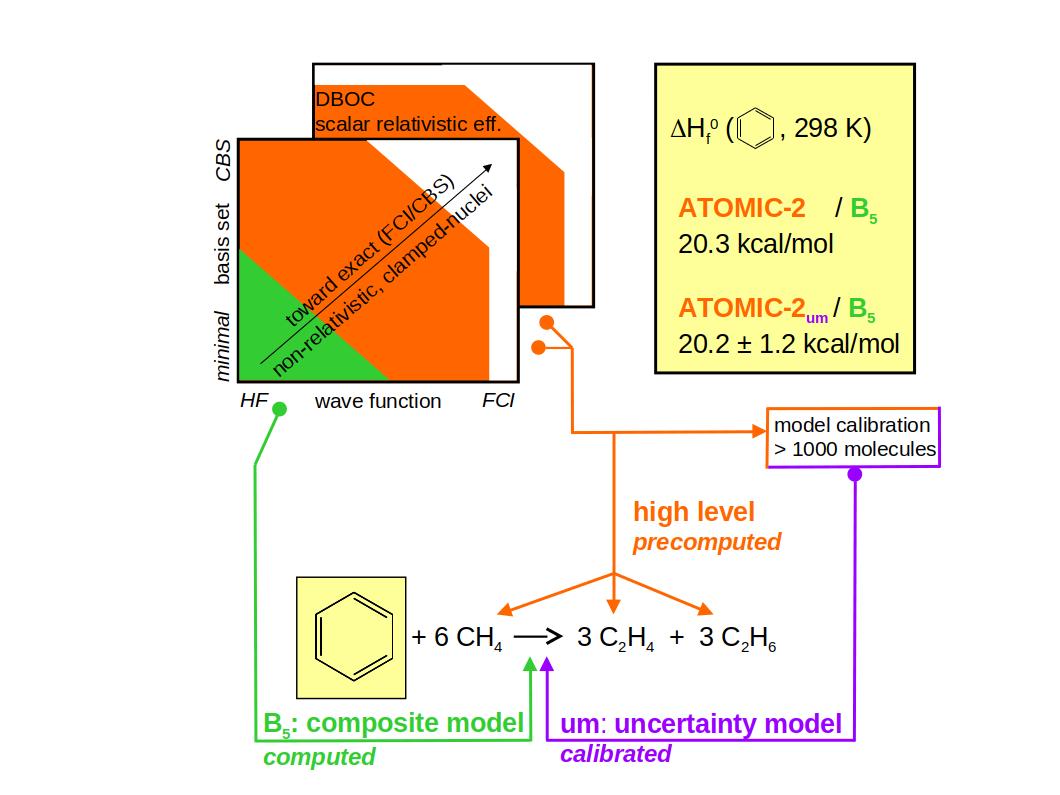
Ab initio thermochemistry: The ATOMIC-2 protocol with estimates of error and uncertainty |
ATOMIC-2 is the latest version of the ATOMIC protocol that implements Pople's
concept of bond separation reactions to reduce the error of midlevel ab initio
approaches in calculations of atomization energies and enthalpies of formation.
The new protocol focuses on computational efficiency and increased accuracy, and
it supplies an estimate of the uncertainty, tailored to size and type of the molecule.
|
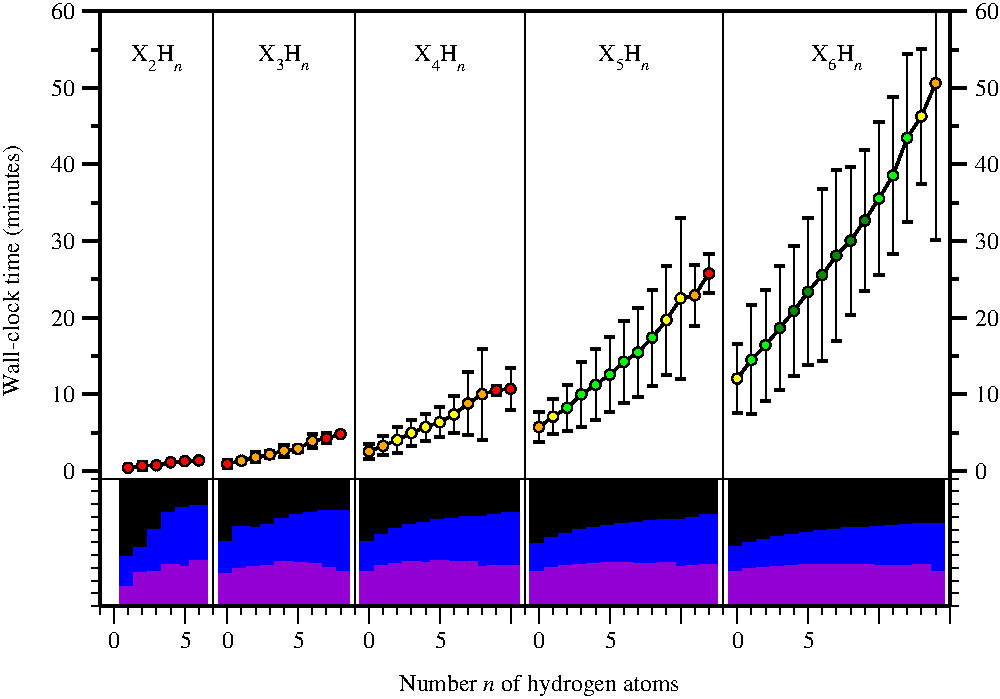
Computational efficiency.
In large benchmarks (> 200'000 molecules) ATOMIC-2 / B5 has been demonstrated to be computationally
very efficient. Molecules with up to six non-hydrogen atoms and any number of hydrogen atoms take less than
an hour to finish, often just a few minutes, using single cores of standard processors.
Yet bias-corrected ATOMIC-2um / B5 achieves an accuracy better than popular G4 theory. |
|
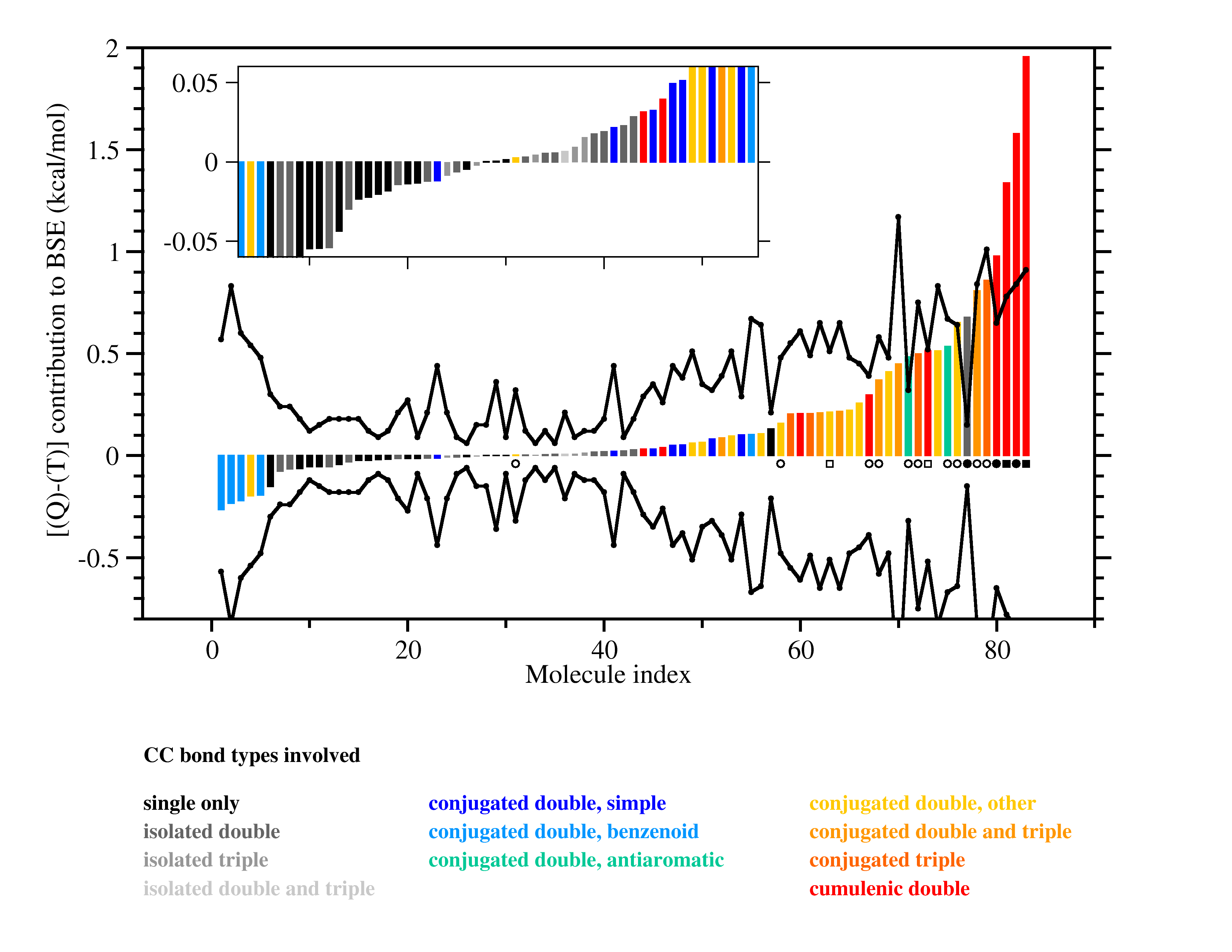
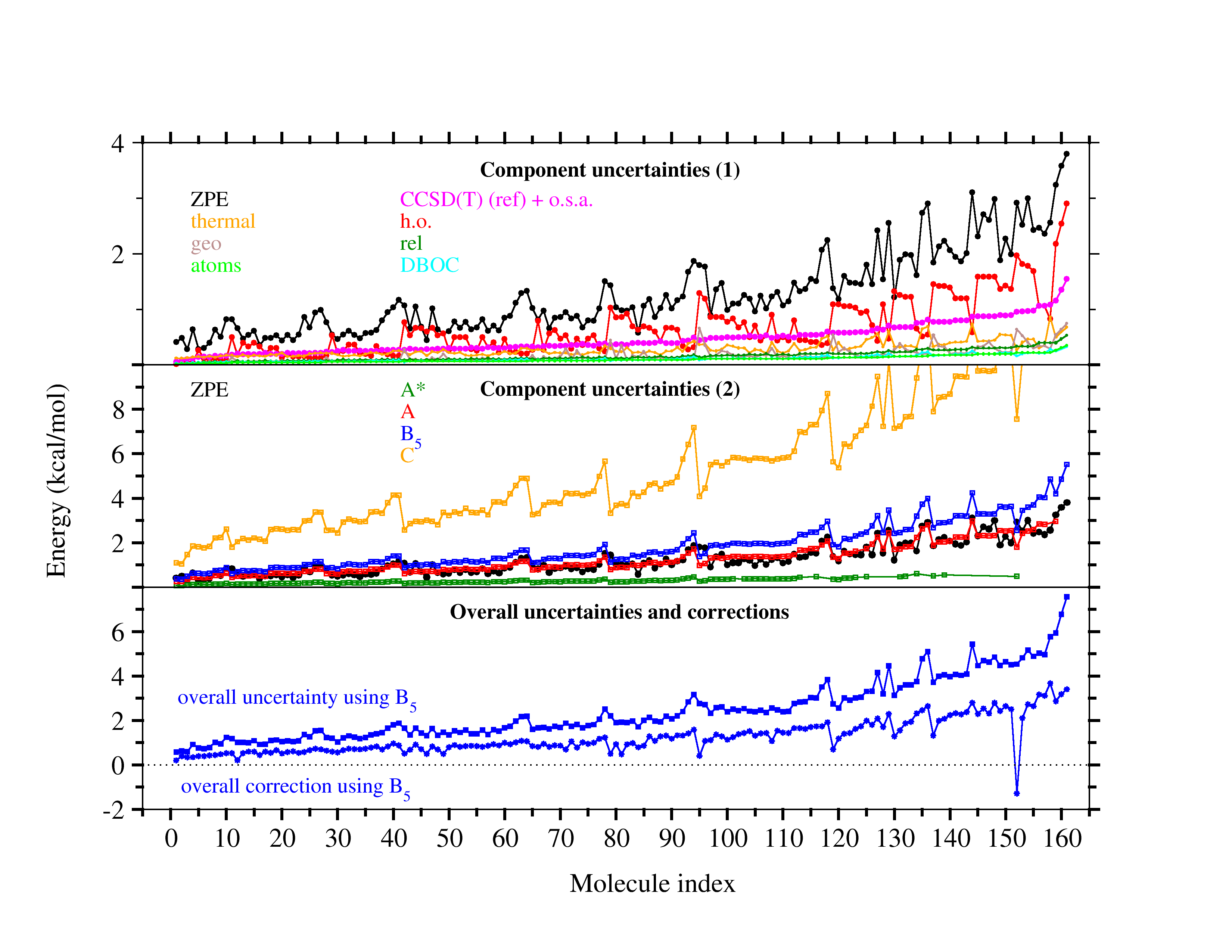
Ab initio thermochemistry: Estimates of error and uncertainty (1) |
In experimental thermochemistry it is accepted standard to
report results together with uncertainties, usually taken to be
intervals of 95% confidence. Theoretical approaches are rarely
designed to provide confidence intervals.
|
The graphic above shows that post-CCSD(T) effects can be quite sizeable, but
it also indicates that simple uncertainty estimates (black lines) manage to
cover most of the systems. A quality criterion computed from the T1 diagnostic
warns of cases for which the error estimate may be unreliable (black circles).
Other sources of error are studied as well, including the limited accuracy of
complete-basis-set extrapolations of CCSD(T) and of computed relativistic effects and
diagonal Born-Oppenheimer corrections.
|
|
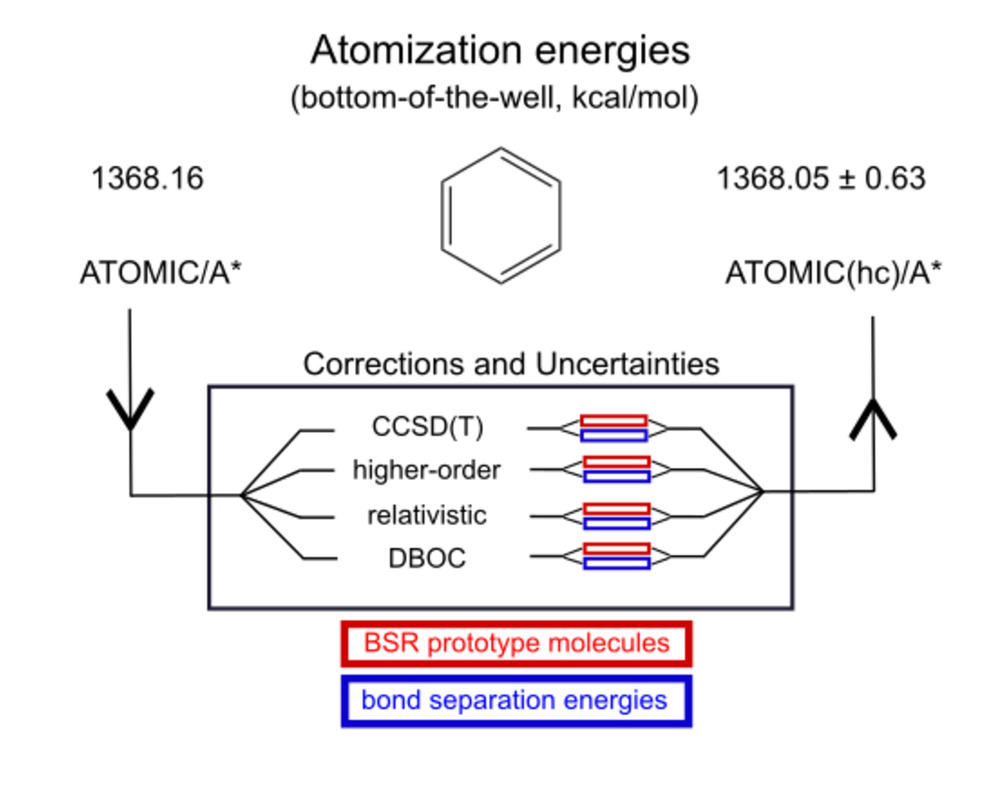
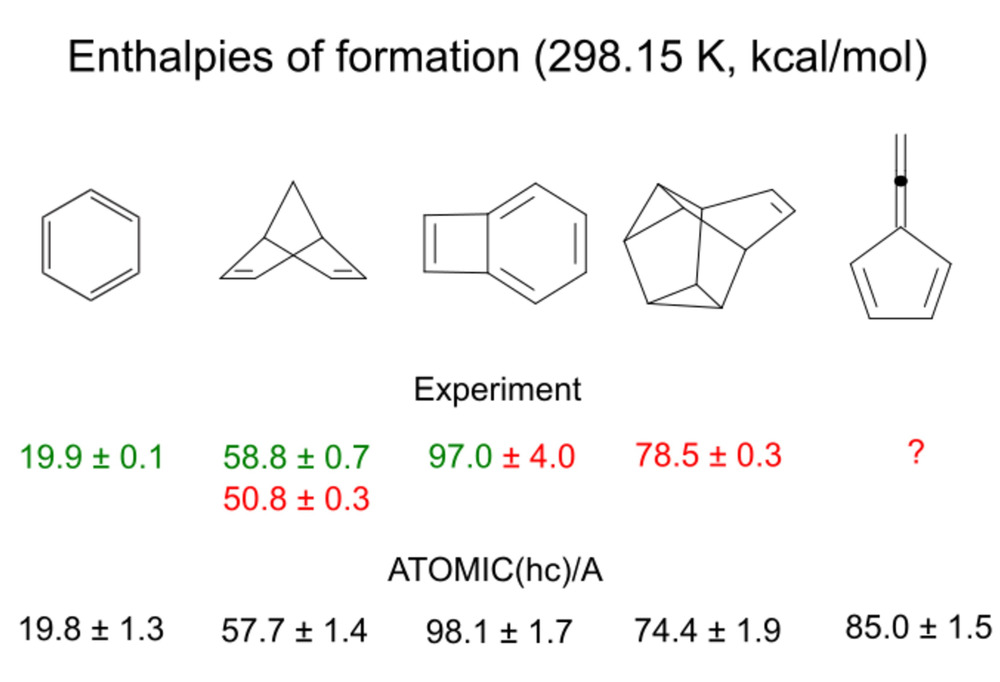
Ab initio thermochemistry: Estimates of error and uncertainty (2) |
In an extension to the ATOMIC(hc) model for bottom-of-the-well
atomization energies, we have also studied errors and uncertainties for
the remaining components necessary to evaluate
enthalpies of formation.
|
The evaluation of ZPEs from scaled harmonic frequencies (black in top graph) expectedly
emerges as the leading source of uncertainty if highly accurate composite models are
used to treat the electronic problem (such as A*, green, vs ZPE, black, in center graph),
but uncertainties are usually balanced with those from computationally more attractive
B level models (such as B5, blue, center graph) to estimate the CBS limit of CCSD(T).
|
|
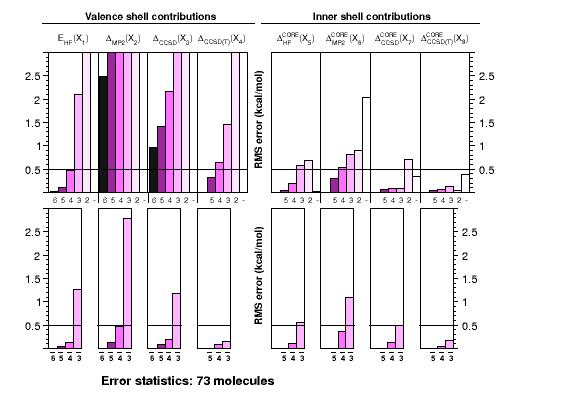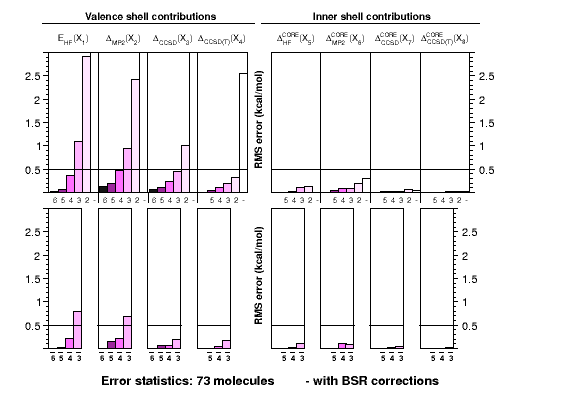
Ab initio thermochemistry: ATOMIC protocol |
The ATOMIC approach
was developed with the needs in mind that are posed by the
calibration of modern approximate models of quantum
chemistry, such as semiempirical methods. It is a robust
and computationally efficient approach to otherwise dauntingly
expensive calculations of atomization energies. The graph shows
how the use of bond separation reactions (BSRs) helps to reduce
errors in each of the components contributing to the CCSD(T)(full)
atomization energy at the complete-basis set limit. Each single
chart shows RMS errors for a particular component as function of
the basis-set cardinal number, without (top) or with (bottom)
extrapolation. In practice only small basis-set calculations
are feasible for larger systems.
|
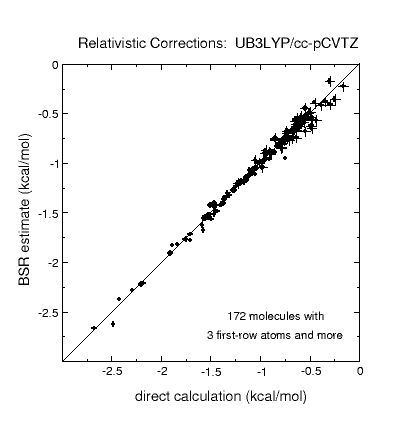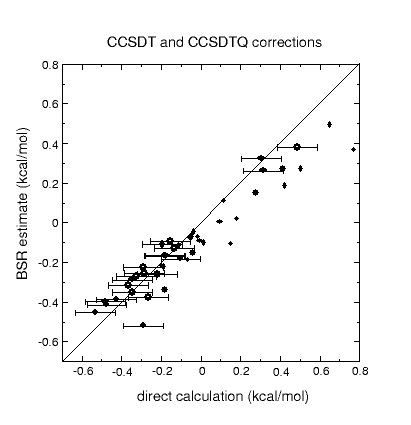
Corrections to atomization energies beyond the CCSD(T) level of theory are
estimated from thermoneutral BSRs. This simplification renders the calculation
of these corrections a trivial task of summing up bond increments. Such an
approach is astonishingly accurate for scalar relativistic corrections and
works reasonably well even for CCSDTQ-CCSD(T) corrections.
|
|
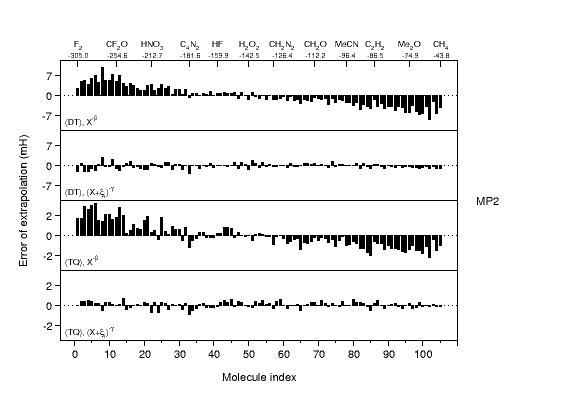
Ab initio thermochemistry: CBS extrapolation |
The development of accurate
extrapolation formulas for electron correlation energies is
an important field in ab initio thermochemistry. Electron correlation
energies are known to converge slowly to the complete basis set
limit, and finite basis set calculations will thus carry substantial
error. On the other hand, computational restraints usually force one
to resort to small basis-set calculations. The graph compares
residual errors for our newly developed and theoretically
well-motivated extrapolation formula (2nd and 4th panel) to those of
the best alternative formulations (1st and 3rd panels). The large
improvement is expected to have a significant impact on producing
reliable ab initio reference data for the calibration of empirical
and semiempirical potentials. |
|
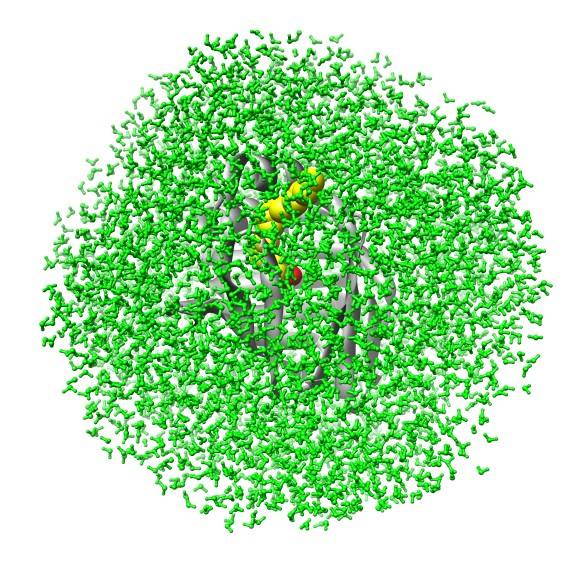
Biomolecular simulation: Algorithms for trajectory analysis |
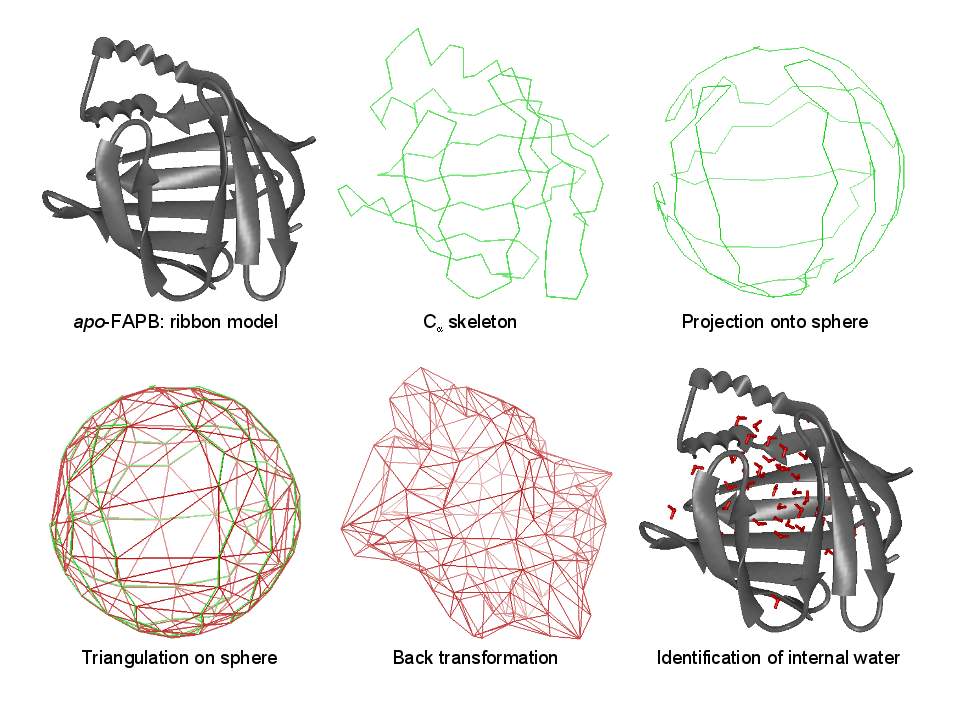
A simulation of FABP in
water. The protein carries a large water-filled cavity in the
interior. How can we extract this internal water?
|
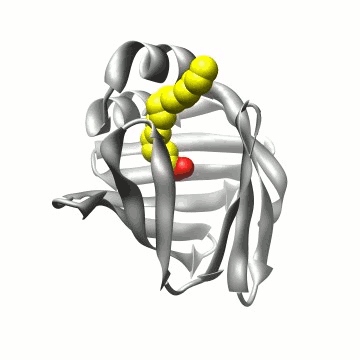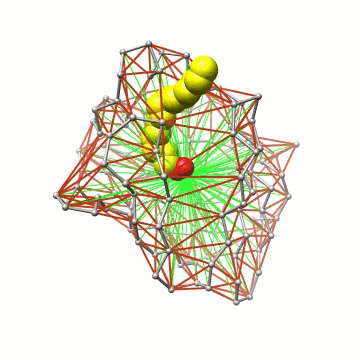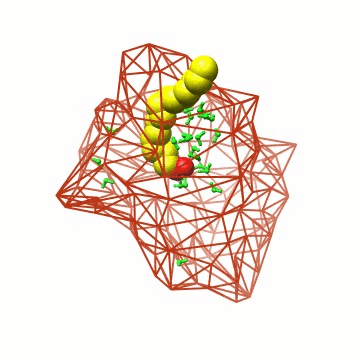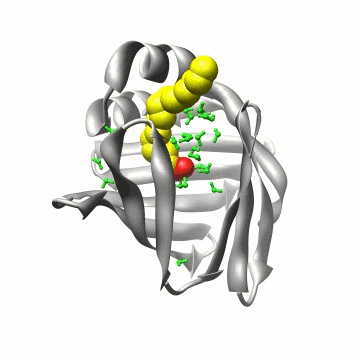
Essentially, we represent the protein barrel by its Cα skeleton, triangulate it, and determine all water molecules inside the polyhedron. |
Some details on the algorithm. The triangulation exploits locality and can thus be performed in linear time. |
|
Biomolecular simulation: FABP |
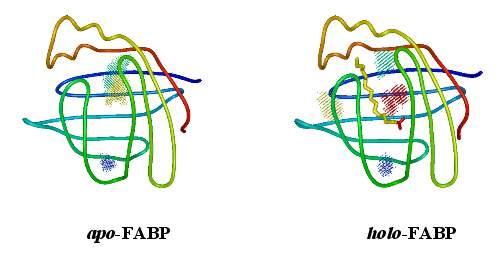
Using the above algorithm, we may
analyze the entire trajectory
and identify the distribution of 3 (apo) and 4 (holo)
water molecules which in NMR experiments have been found to be
particularly immobile,
|
or analyze the entire interior water density |
or even time-resolved interaction potentials with other water, with protein residues, and with the ligand to improve our understanding of the internal water dynamics. |
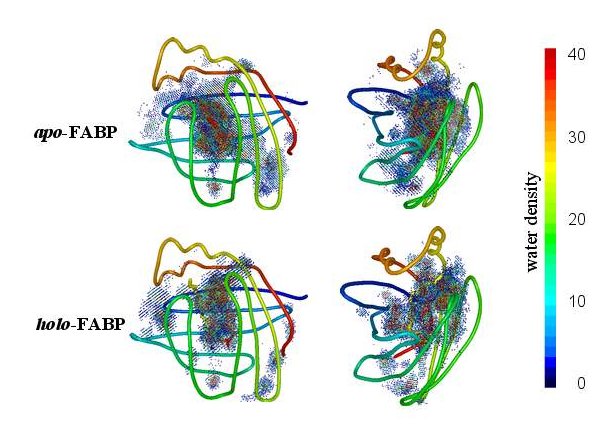
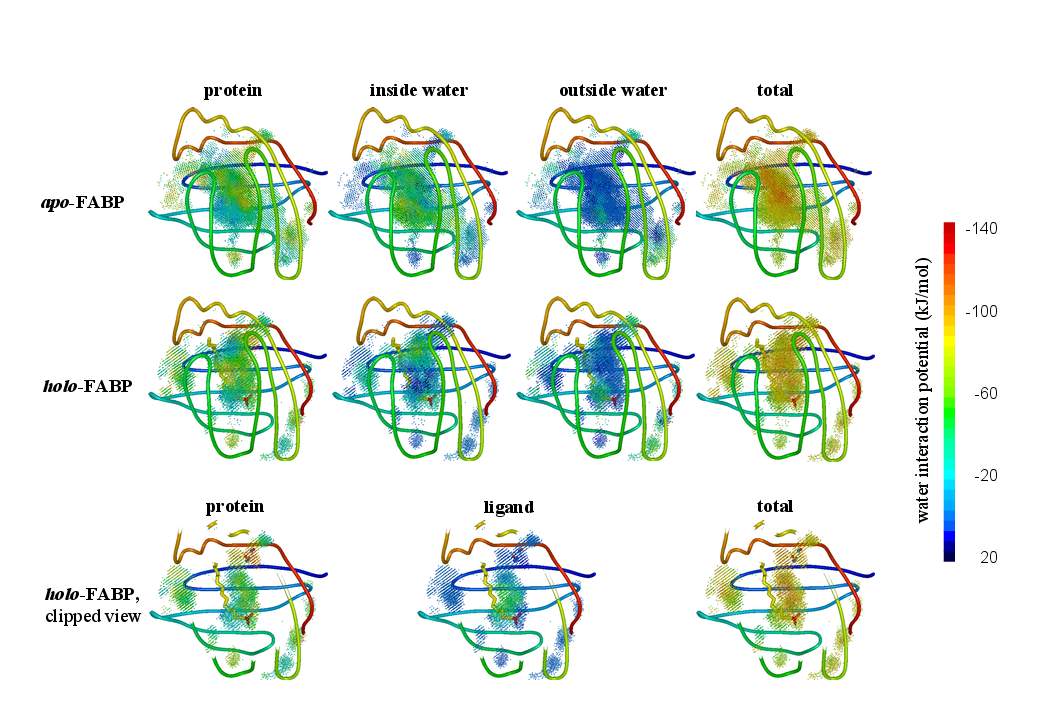
|
Biomolecular simulation: Carbopeptoids |
Carbopeptoids are homooligomers of sugar-containing peptides,
and they serve as rigidified peptide models with potential
applications as drugs that block protein-protein interactions
and inhibit enzyme catalysis.
|
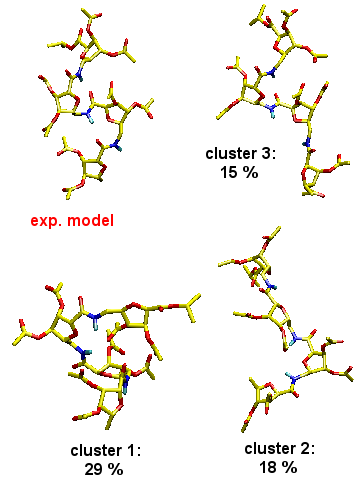
MD simulations
reproduce experimentally (NOE) derived distance constraints.
Cluster analyses of MD trajectories demonstrates, however,
that the experimentally postulated helical structure is only
one of several dominating structural motifs comprising the
entire ensemble, and that the unfolded state is in fact not
structureless. Such insight is hard if not impossible to obtain
from experiment alone.
|
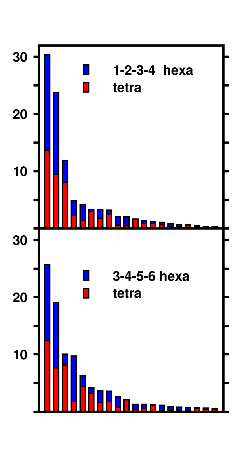
Cluster analysis combining the ensembles of the tetrapeptide and equally long blocks of the hexapeptide demonstrates the repetition of structural motifs in longer peptide chains, a result, that was postulated in experimental studies. Note the "overlapping" (blue/red) clusters in the graph. |
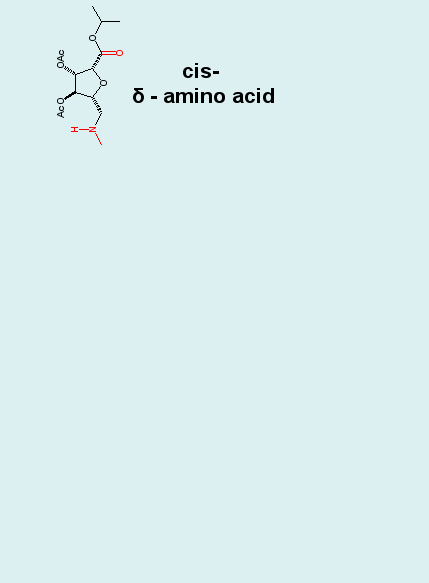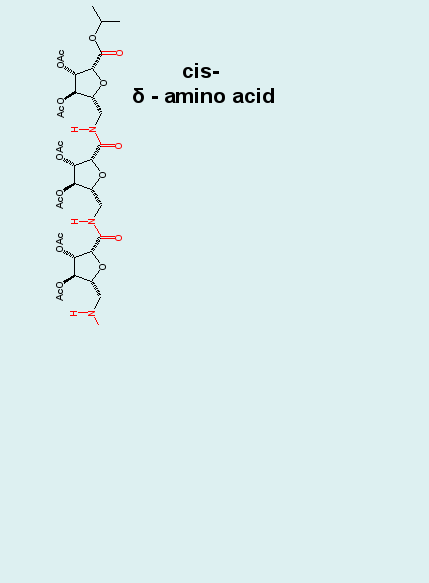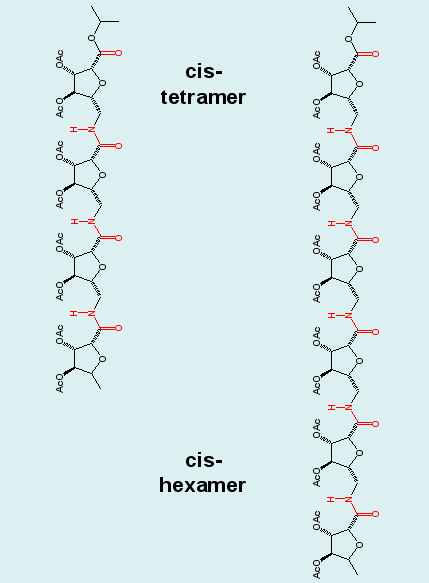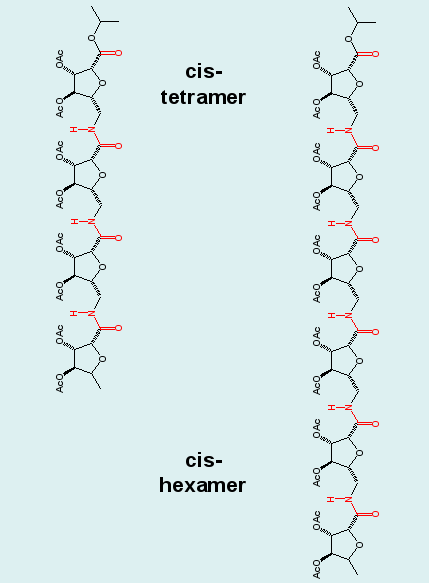
|
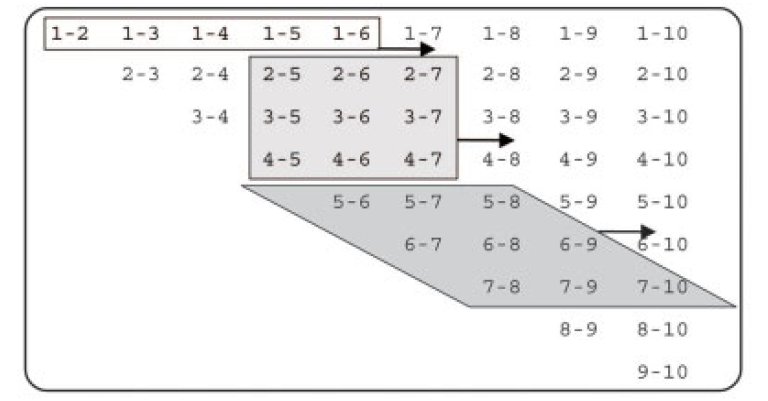
MD simulation software: Pair list algorithms |
MD simulations often apply a distance-cutoff for pair potentials,
and the scan of the atom pair matrix is one of the very
time-critical parts of such an MD simulation. While linear-scaling
grid-cell techniques become efficient for very large system sizes,
improved double-loop algorithms are beneficial for intermediate
sizes often considered in current-day simulations.
|
Here we take advantage of the fast processor cache found in modern CPUs and replace the row-wise atom pair scan (unshaded) by a window scan (shaded) which can process a number of pairs that scales quadratically, rather than linearly, with the number of atoms loaded into cache memory. The triangular atom-pair matrix may be reordered to become rectangular, in which case all rhombic windows become quadratic. |

|
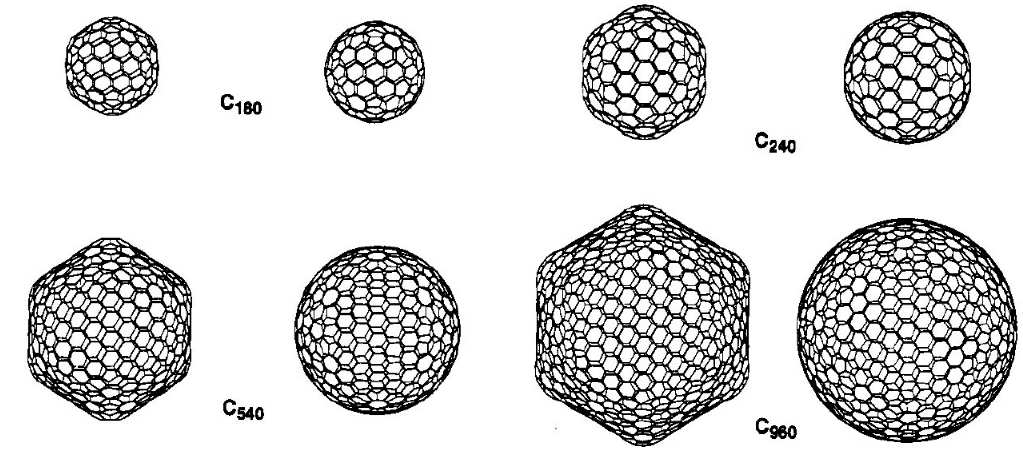
Giant fullerenes |
Fullerenes were discovered in the mid-80's and have attracted
a lot of attention as new allotropes of carbon. The prototype
buckminsterfullerene, C60, is spherical due to its
high symmetry.
|
Despite earlier claims, however, our
semiempirical calculations
have shown that larger fullerenes of icosahedral symmetry
prefer facetted over spherical shapes. These results were
confirmed by more rigorous
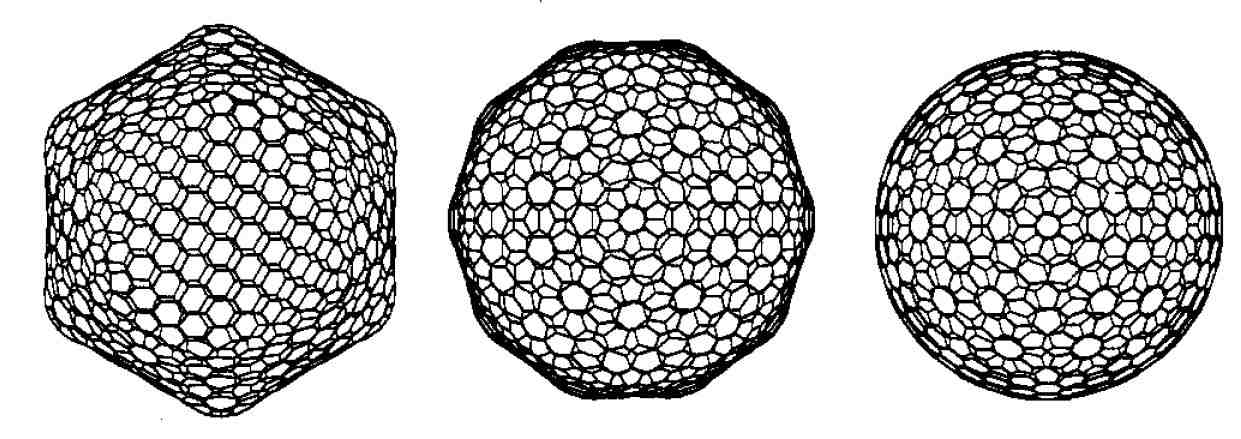
density functional calculations . The picture above shows
the facetted form of C960 from two different
perspectives and a hypothetical spherical alternative. |