PUBLICATIONS
Dirk Bakowies
|
Bakowies, D. ,
"
ATOMIC-2 protocol for thermochemistry
"
J. Chem. Theory Comput. 2022, 18, 4142-4163.
Journal Page
Supplementary Material (PDF)
Supplementary Material (ZIP)
Abstracts off
Computer code available.
Bakowies D.,
"Ab initio thermochemistry with ATOMIC-2"
DOI: 10.5281/zenodo.5780172
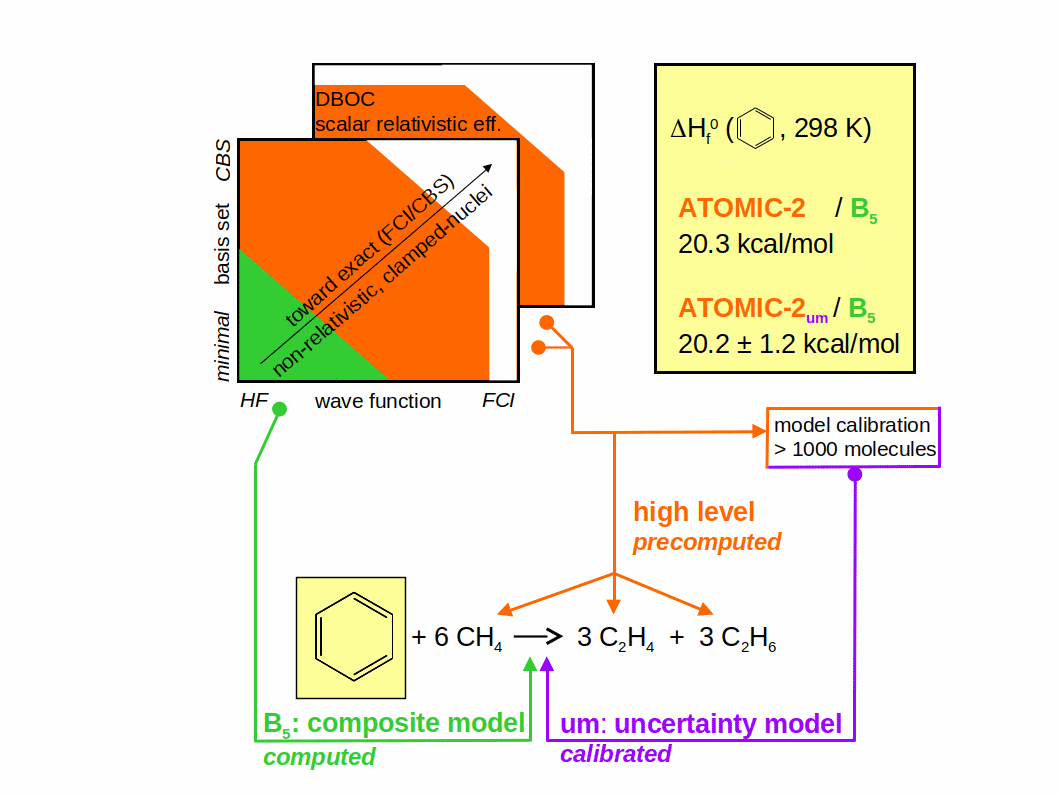
ATOMIC is a midlevel thermochemistry protocol that uses Pople's concept of bond separation reactions (BSRs) as theoretical framework to reduce computational demands in the evaluation of atomization energies and enthalpies of formation. Various composite models are available that approximate bond separation energies at the complete basis set limit of all-electron CCSD(T), each balancing computational cost with achievable accuracy. Evaluated energies are then combined with very high-level, precomputed atomization energies of all auxiliary molecules appearing in the BSR to obtain the atomization energy of the molecule under study. ATOMIC-2 is a new version of the protocol that retains the overall concept and all previously defined composite models, but improves on ATOMIC-1 in various other ways: Geometry optimization and zero-point-energy evaluation are performed at the density functional level (PBE0-D3/6-311G(d)), which shows significant computational savings and better accuracy than the previously employed RI-MP2/cc-pVTZ. The BSR framework is improved, using more accurate CBS extrapolations toward the Full CI limit for the atomization energies of all auxiliary molecules. Finally, and most importantly, an error and uncertainty model termed ATOMIC-2_um is added that estimates average bias and uncertainty for each of the atomization energy contributions that arise from simplified treatment of some contributions to bond separation energies (CCSD(T)) and neglect of others (such as higher order, scalar relativistic or diagonal Born-Oppenheimer corrections) or from residual error in the energies of auxiliary molecules. Large and diverse benchmarks including up to 1179 molecules are used to evaluate necessary reference data and to correlate observed error for each of the contributions with appropriate proxies that are available without additional quantum-chemical calculation for a particular molecule and represent its size and type. The implementation of ATOMIC-2 considers neutral, closed-shell molecules containing H, C, N, O, and F atoms; compared to ATOMIC-1 the framework has been extended to cover a few challenging but rare bond topologies. In comparison to highly accurate reference data for 184 molecules taken from the ATcT database (V. 1.122r), regular ATOMIC-2 shows noticable underbinding, but the bias-corrected protocol ATOMIC-2_um is found to be more accurate than either ATOMIC-1 or standard Gaussian-4 theory, and the uncertainty model is consistent with statistics of actually observed errors. Problems arising from ambiguous or challenging Lewis-valence structures defining BSRs are discussed, and computational efficiency is demonstrated. Computer code is made available to perform ATOMIC-2um analyses.
|
|
Bakowies, D.; von Lilienfeld, O. A. ,
"
Density functional geometries and zero-point energies in ab initio thermochemical treatments of compounds with first-row atoms (H, C, N, O, F)
"
J. Chem. Theory Comput. 2021, 17(8), 4872-4890.
Journal Page
Supplementary Material (PDF)
Supplementary Material (ZIP)
Abstracts off
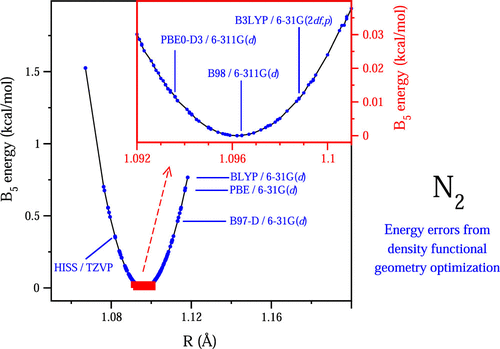
Density functionals are often used in ab initio thermochemistry to provide optimized geometries for single-point evaluations at high level and to supply estimates of anharmonic zero-point energies (ZPEs). Their use is motivated by relatively high accuracy at modest computational expense, but a thorough assessment of geometry-related error seems to be lacking. We have benchmarked 53 density functionals, focusing on approximations of the first four rungs and on relatively small basis sets for computational efficiency. Optimized geometries of 279 neutral first-row molecules (H, C, N, O, F) are judged by energy penalties relative to the best available geometries, using the composite model ATOMIC / B5 as energy probe. Only hybrid functionals provide good accuracy with root mean square errors around 0.1 kcal/mol and maximum errors below 1.0 kcal/mol, but not all of them do. Conspicuously, first-generation hybrids with few or no empirical parameters tend to perform better than highly parameterized ones. A number of them show good accuracy already with small basis sets (6-31G(d), 6-311G(d)). As is standard practice, anharmonic ZPEs are estimated from scaled harmonic values. Statistics of the latter show less performance variation among functionals than observed for geometry-related error, but they also indicate that ZPE error will generally dominate. We have selected PBE0-D3/6-311G(d) for the next version of the ATOMIC protocol (ATOMIC-2) and studied it in more detail. Empirical expressions have been calibrated to estimate bias corrections and 95% uncertainty intervals for both geometry-related error and scaled ZPEs.
|
|
Bakowies, D. ,
"
Estimating systematic error and uncertainty in ab initio thermochemistry: II. ATOMIC(hc) enthalpies of formation for a large set of hydrocarbons
"
J. Chem. Theory Comput. 2020, 16(1), 399-426.
Journal Page
Supplementary Material
Abstracts off
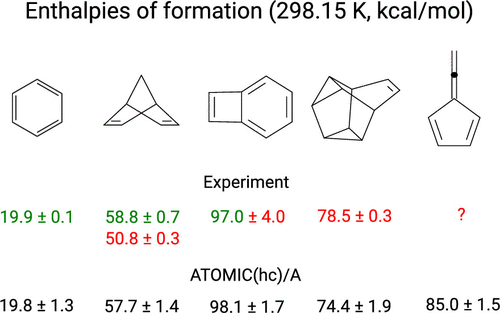
ATOMIC is a thermochemistry protocol geared toward larger molecules with first-row atoms. It implements Pople's concept of bond separation reactions in an ab initio fashion and so enhances the accuracy of midlevel composite models for atomization energies. Recently we have introduced ATOMIC(hc), a model for applications to hydrocarbons, that estimates bias and uncertainty for each of the components contributing to the ATOMIC bottom-of-the-well atomization energy (Bakowies, D. J. Chem. Theory Comput. 2019, 15, 5230-5251). Here we scrutinize the remaining components of the ATOMIC protocol, including midlevel composite models to approximate the complete-basis set (CBS) limit of CCSD(T) as well as zero-point energies (ZPEs) and thermal enthalpy increments that are evaluated from scaled harmonic MP2 frequencies. Potential errors relating to imperfections in MP2 geometries and ZPEs are estimated using auxiliary information obtained from geometry optimizations and frequency calculations at the density functional (B3LYP) level. Overall corrections to and uncertainties of enthalpies of formation are obtained from summation and error propagation, respectively. The error and uncertainty model is validated with accurate data from the Active Thermochemical Tables (ATcT) and compared to earlier statistical assessments for the G3/99 benchmark. The proposed model is a welcome alternative to statistical assessment, first because it does not depend on comparison with experiment, second because it recognizes the expected scaling of error with system size, and third because it provides a detailed account of the importance of various contributions to overall error and uncertainty. The evaluation of ZPEs from scaled harmonic frequencies expectedly emerges as the leading source of uncertainty if highly accurate composite models are used to treat the electronic problem, but uncertainties are usually balanced with those arising from computationally more attractive B level (B1...B6) models to estimate the CBS limit of CCSD(T). ATOMIC(hc) enthalpies of formation, complete with uncertainty estimates, are reported for 161 hydrocarbons ranging in size from methane (CH4) to [8]circulene (C32H16) and tetra-tert-butyltetrahedrane (C20H36). Experimental data are available for 127 molecules but cannot be reconciled with theory in 37 cases. Theory helps to identify the more accurate among conflicting experimental values in 11 cases and emerges as a valuable complement to experiment also for larger molecules, provided that fair estimates of uncertainty are available.
|
|
Bakowies, D. ,
"
Estimating systematic error and uncertainty in ab initio thermochemistry. I. Atomization energies of hydrocarbons in the ATOMIC(hc) protocol
"
J. Chem. Theory Comput. 2019, 15(10), 5230-5251.
Journal Page
Supplementary Material
Abstracts off
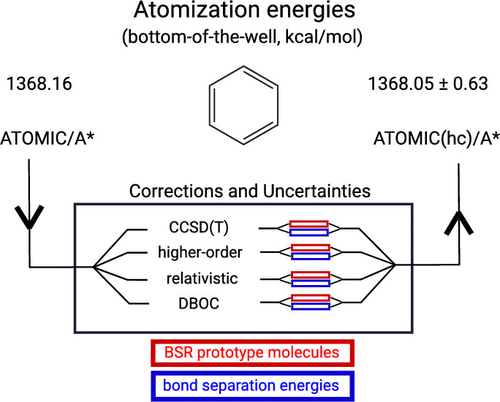
Research in ab initio quantum chemistry has produced an increasing number of thermochemistry protocols, serving different needs from benchmark-level accuracy for small molecules to "chemical accuracy" for larger molecules. While in experimental thermochemistry it is accepted standard to report results complete with intervals of 95% confidence, so far few of the most advanced theoretical approaches have followed suit, based on either statistical comparison to well-established experimental data or careful assessment of high level theoretical results for individual molecules. Here we report on the development of intrinsic uncertainty estimates for the ATOMIC protocol in applications to hydrocarbons. ATOMIC is a theoretical procedure geared toward larger molecules and based on the ab initio implementation of bond separation reactions (BSRs) to reduce errors of midlevel composite approaches. Each of the components contributing to the bottom-of-the-well atomization energy (EA,e) is scrutinized for possible error by comparison to a large number of very high-level results, including complete-basis-set estimates of CCSDT(Q) bond separation energies for 83 hydrocarbons up to the size of naphthalene. Some of the observations are the following: Post-CCSD(T) effects are sizable even for saturated aliphatic compounds but well-represented in a BSR model summing over bond contributions, while conjugated systems pose more problems. Another significant source of error is the complete-basis-set extrapolation of all-electron CCSD(T) contributions, which still carries an uncertainty of a few tenths of a kcal/mol for midsize molecules like benzene, even if based on large basis-set calculations (cc-pCV5Z, cc-pCV6Z). Scalar relativistic terms and diagonal Born-Oppenheimer corrections are of less concern, the former because they are well represented in a BSR model and the latter because they are small in general. Observations are cast into simple expressions that separate obvious bias from nonsystematic error, formulating the former as correction to and the latter as uncertainty of an ATOMIC result. The updated protocol, complete with uncertainties and termed ATOMIC(hc) ("hc" for hydrocarbons), is validated in comparisons with both experimental data from the Active Thermochemical Tables and high-level theoretical data generated in this work. Analysis of lower-level ATOMIC models and of further components needed to convert EA,e into enthalpies of formation will be reported separately.
|
|
Tahchieva, D. N.; Bakowies, D.; Ramakrishnan R.; von Lilienfeld, O. A. ,
"
Torsional potentials of glyoxal, oxalyl halides, and their thiocarbonyl derivatives: Challenges for popular density functional approximations
"
J. Chem. Theory Comput. 2018, 14(9), 4806-4817.
Citing Articles (ISI)
Journal Page
Supplementary Material
1 /
2 /
3 /
Abstracts off
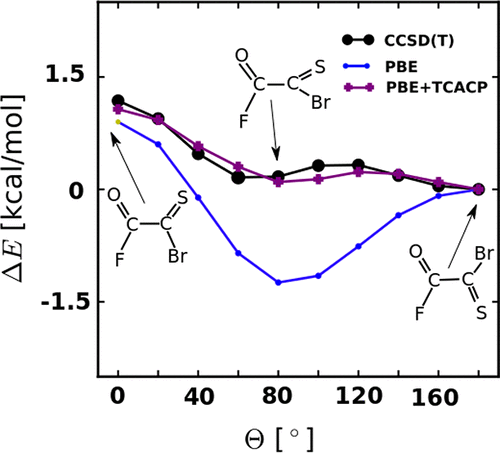
The reliability of popular density functionals was studied for the description of torsional profiles of 36 molecules: glyoxal, oxalyl halides, and their thiocarbonyl derivatives. HF and 18 functionals of varying complexity, from local density to range-separated hybrid approximations and double-hybrid, have been considered and benchmarked against CCSD(T)-level rotational profiles. For molecules containing heavy halogens, most functionals fail to reproduce barrier heights accurately and a number of functionals introduce spurious minima. Dispersion corrections show no improvement. Calibrated torsion-corrected atom-centered potentials rectify the shortcomings of PBE and also improve on sigma-hole based intermolecular binding in dimers and crystals.
|
|
Bakowies, D. ,
"
Simplified wave function models in thermochemical protocols based on
bond separation reactions
"
J. Phys. Chem. A 2014, 118(50), 11811-11827.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
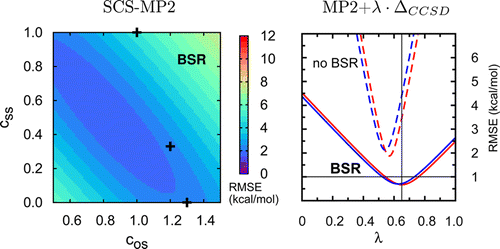
The ATOMIC protocol is a quantum-chemical thermochemistry protocol designed to obtain accurate atomization energies and derived heats of formation. It reduces errors of computationally tractable composite schemes through the use of bond separation reactions, which are implemented in a consistent ab initio framework. The present work explores possible simplification of previously introduced ATOMIC models. While coupled cluster calculations with singles and doubles excitations and perturbational treatments of connected triples excitations [CCSD(T)] are still required for high accuracy, basis-set truncations are possible in the CCSD-MP2 and CCSD(T)-CCSD components. The resulting models B4, B5, and B6 show root-mean-square (RMS) errors of only 0.21 to 0.46 kcal/mol for the AE set, which is a benchmark comprising complete-basis-set CCSD(T)(full) atomization energies of 73 neutral, closed-shell molecules composed of H, C, N, O, and F atoms. The evaluation of connected triples excitations can be avoided at medium levels of accuracy if the complete-basis-set MP2 energy is augmented with an empirically calibrated fraction of the difference between MP3 (or CCSD) and MP2 energies, calculated with small basis sets. The corresponding EMP3 and ECCSD models show RMS errors of 1.01 and 0.70 kcal/mol, respectively. Spin-component scaling is an option to rely entirely on the MP2 level of theory and still cut the RMS error of 4.38 kcal/mol by roughly a factor of 2 and achieve an accuracy comparable to accurate density functionals, such as M05-2X. The proposed new models are additionally tested with the HOF benchmark, a subset of G3/99 heats of formation that includes only neutral closed-shell molecules composed of H, C, N, O, and F atoms. The assessment shows that a number of experimental reference values are in error and should be replaced with more recent data. Results obtained with the new models are compared to original HOF (G3/99) reference data, to updated reference data, and to accurate ATOMIC/A theoretical data.
|
|
Bakowies, D. ,
"
Assessment of density functional theory for thermochemical approaches
based on bond separation reactions
"
J. Phys. Chem. A 2013, 117(1), 228-243.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
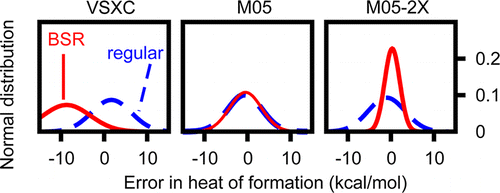
The recently proposed ATOMIC protocol is a fully ab initio
thermochemical protocol that rests upon the concept of bond
separation reactions (BSRs) to correct for systematic errors
of composite wave function approaches. It achieves high
accuracy for atomization energies and derived heats of
formation if basis-set requirements for all contributing
components are balanced carefully. The present work explores
the potential of density functionals as possible replacements
of composite wave function approaches in the ATOMIC protocol.
Twenty density functionals are examined for their accuracy
in thermochemical predictions based on calculated bond-
separation energies and precomputed high-level data for the
small parent molecules entering BSRs. The best density
functionals outperform CCSD (coupled cluster with singles
and doubles excitations), but none reaches the accuracy
of well-balanced composite wave function approaches that
consider quasiperturbational connected triples excitations
at least with small basis sets. Some functionals show unexpected
problems with bond separation reactions and are analyzed
further with a model of empirically calibrated bond
additivity corrections. Finally, the benefit of adding
empirical dispersion terms to common density functionals
is analyzed in the context of BSR-corrected thermochemistry.
|
|
Bakowies, D. ,
"
Ab initio thermochemistry with high-level isodesmic corrections:
Validation of the ATOMIC protocol for a large set of compounds with
first-row atoms (H, C, N, O, F)
"
J. Phys. Chem. A 2009, 113(43), 11517-11534.
Citing Articles (ISI)
Journal Page
Abstracts off
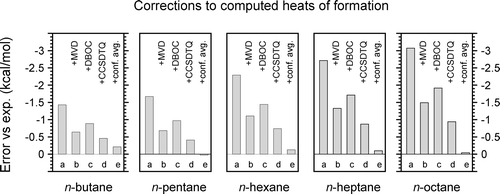
The recently proposed ATOMIC protocol is a fully ab initio
thermochemical approach designed to provide accurate
atomization energies for molecules with well-defined
valence structures. It makes consistent use of the concept
of bond-separation reactions to supply high-level precomputed
bond increments which correct for errors of lower-level models.
The present work extends the approach to the calculation of
standard heats of formation and validates it by comparison to
experimental and benchmark level ab initio data reported in the
literature. Standard heats of formation (298 K) have been compiled
for a large sample of 173 neutral molecules containing hydrogen
and first-row atoms (C, N, O, F), resorting to several previous
compilations and to the original experimental literature. Statistical
evaluation shows that the simplest implementation of the ATOMIC
protocol (composite model C) achieves an accuracy comparable to
the popular Gaussian-3 approach and that composite models A and B
perform better. Chemical accuracy (1 - 2 kcal/mol) is normally achieved
even for larger systems with about 10 non-hydrogen atoms and for
systems with charge-separated valence structures, bearing testimony
to the robustness of the bond-separation reaction model. Effects of
conformational averaging have been examined in detail for the series
of n-alkanes, and our most refined composite model A reproduces
experimental heats of formation quantitatively, provided that
conformational averaging is properly accounted for. Several
cases of larger discrepancy with respect to experiment are
discussed, and potential weaknesses of the approach are identified.
|
|
Bakowies, D. ,
"
Ab initio thermochemistry using optimal-balance models with isodesmic corrections: The ATOMIC protocol
"
J. Chem. Phys. 2009, 130, 144113/1-21.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
Free Download Copyright Notice: Copyright (2009) American Institute of Physics. This article may be downloaded for personal use only. Any other use requires prior permission of the author and the American Institute of Physics. The article appeared in the Journal of Chemical Physics (citation above) and may be found at the journal webpage .
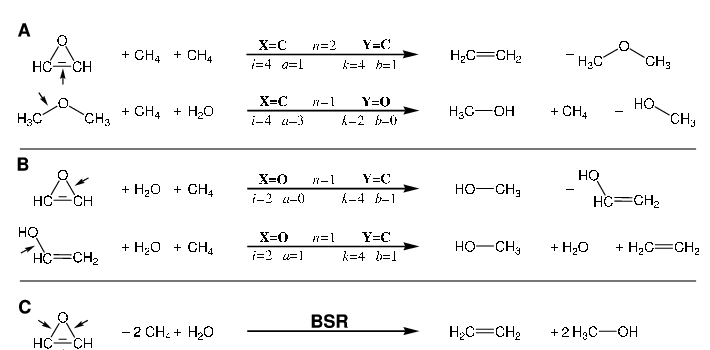
A theoretical composite approach, termed ATOMIC for "Ab initio
Thermochemistry using Optimal-balance Models with Isodesmic Corrections",
is introduced for the calculation of molecular atomization energies and
enthalpies of formation. Care is taken to achieve optimal balance in
accuracy and cost between the various components contributing to high-level
estimates of the fully correlated energy at the infinite-basis set limit.
To this end, the energy at the coupled-cluster level of theory including
single, double, and quasi-perturbational triple excitations is decomposed
into Hartree-Fock, low-order correlation (MP2, CCSD) and connected-triples
contributions, and into valence-shell and core contributions. Statistical
analyses for 73 representative neutral closed-shell molecules containing
hydrogen and at least three first-row atoms (CNOF) are used to devise
basis set and extrapolation requirements for each of the eight components
to maintain a given level of accuracy. Pople's concept of bond-separation
reactions is implemented in an ab initio framework, providing for a
complete set of high-level precomputed isodesmic corrections which can
be used for any molecule for which a valence structure can be drawn. Use
of these corrections is shown to lower basis set requirements dramatically
for each of the eight components of the composite model. A hierarchy of
three levels is suggested for isodesmically corrected composite models
which reproduce atomization energies at the reference level of theory to
within 0.1 kcal/mol (A), 0.3 kcal/mol (B), and 1 kcal/mol (C). Large-scale
statistical analysis shows that corrections beyond the CCSD(T) reference
level of theory, including coupled cluster theory with fully relaxed
connected triple and quadruple excitations, first-order relativistic and
diagonal Born-Oppenheimer corrections can normally be dealt with using a
greatly simplified model that assumes thermoneutral bond-separation reactions
and that reduces the estimate of these corrections to the simple task of
adding up bond increments. Preliminary validation with experimental
enthalpies of formation, using the subset of neutral closed-shell (HCNOF)
species contained in the the G3/99 test set, indicates that the ATOMIC
protocol performs slightly better than the popular G3 approach. The newly
introduced protocol does not require empirical calibration, however, and it
is still efficient enough to be applied routinely to molecules with
ten or twenty non-hydrogen atoms.
|
|
Bakowies, D. ,
"
Ab initio thermochemistry of large molecules
"
Scientific Report of the Swiss National Supercomputing Centre (2006/2007) 2008, 32-35.
Scientific Report
Abstracts off
Thermochemistry is a branch of thermodynamics
concerned with the energy balance of chemical reactions.
The elements in their standard states define
the universal reference, establishing heats of
formation as the primary quantity relating the heat
content of one compound to that of another.
Experimental access is usually provided through
combustion calorimetry, supplemented by measurements
of heats of vaporization or sublimation.
On the theoretical side, heats of formation may be
obtained from atomization energies, but ab initio
predictions to any useful accuracy (say 1-2 kcal/
mol) have not been possible even for the smallest
chemical systems until about 20 years ago. This is
mainly attributable to the large error incurred if
electron correlation is not dealt with accurately, as
the total atomization of a molecule involves a significant
change in electron correlation. Accurate
treatment, however, requires expensive calculations
using extended basis sets as the electron
correlation energy is known to converge very
slowly with the size of orbital-based expansions.
Our interest in accurate ab initio thermochemical
approaches reflects the need for supplying high-quality
reference data for semiempirical method
development. Traditionally such reference data
have been obtained almost exclusively from experiment,
but a reliable ab initio protocol would offer
several advantages: (a) Heats of formation,
previously used but theoretically not well justified,
can be replaced by more appropriate atomization
energies which are easily available only from calculation.
(b) The accuracy of experimental data is
often hard to quantify, and while many data are
very well established, occasionally large errors do
occur. (c) Experimental data are entirely unavailable
for several important classes of molecules,
and, in particular for biologically relevant model
systems such as peptides. The latter point is of
particular importance as one of the most promising
fields of application for improved semiempirical
methodology is in biochemistry.
|
|
Bakowies, D. ,
"
Accurate extrapolation of electron correlation energies from small basis sets
"
J. Chem. Phys. 2007, 127, 164109/1-12
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
Free Download Copyright Notice: Copyright (2007) American Institute of Physics. This article may be downloaded for personal use only. Any other use requires prior permission of the author and the American Institute of Physics. The article appeared in the Journal of Chemical Physics (citation above) and may be found at the journal webpage .
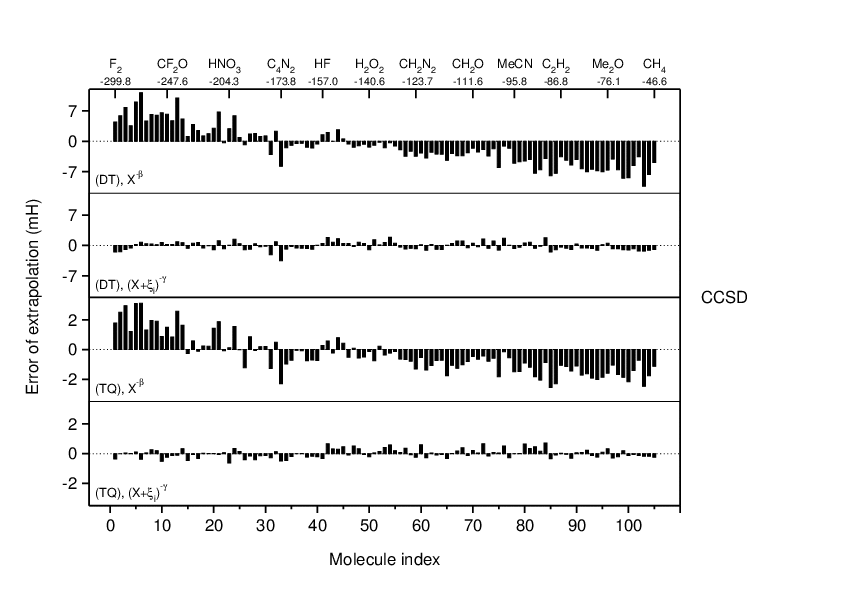
A new two-point scheme is proposed for the extrapolation of electron
correlation energies obtained with small basis sets. Using the series
of correlation-consistent polarized valence basis sets, cc-pVXZ, the
basis set truncation error is expressed as δEX∝(X+ ξi)-γ.
The angular momentum offset ξi captures differences in effective rates
of convergence previously observed for first-row molecules. It is based
on simple electron counts and tends to values close to 0 for hydrogen-rich
compounds and values closer to 1 for pure first-row compounds containing
several electronegative atoms. The formula is motivated theoretically by
the structure of correlation-consistent basis sets which include basis
functions up to angular momentum L=X-1 for hydrogen and helium and up to
L=X for first-row atoms. It contains three parameters which are calibrated
against a large set of 105 reference molecules (H, C, N, O, F) for
extrapolations of MP2 and CCSD valence-shell correlation energies from
double- and triple-zeta (DT) and triple- and quadruple-zeta (TQ) basis
sets. The new model is shown to be three to five times more accurate
than previous two-point schemes using a single parameter, and (TQ)
extrapolations are found to reproduce a small set of available R12
reference data better than even (56) extrapolations using the conventional
asymptotic limit formula δEX∝X -3. Applications to a small
selection of boron compounds and to neon show very satisfactory results
as well. Limitations of the model are discussed.
|
|
Bakowies, D. ,
"
Extrapolation of electron correlation energies to finite and complete basis set targets
"
J. Chem. Phys. 2007, 127, 084105/1-23.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
Free Download Copyright Notice: Copyright (2007) American Institute of Physics. This article may be downloaded for personal use only. Any other use requires prior permission of the author and the American Institute of Physics. The article appeared in the Journal of Chemical Physics (citation above) and may be found at the journal webpage .
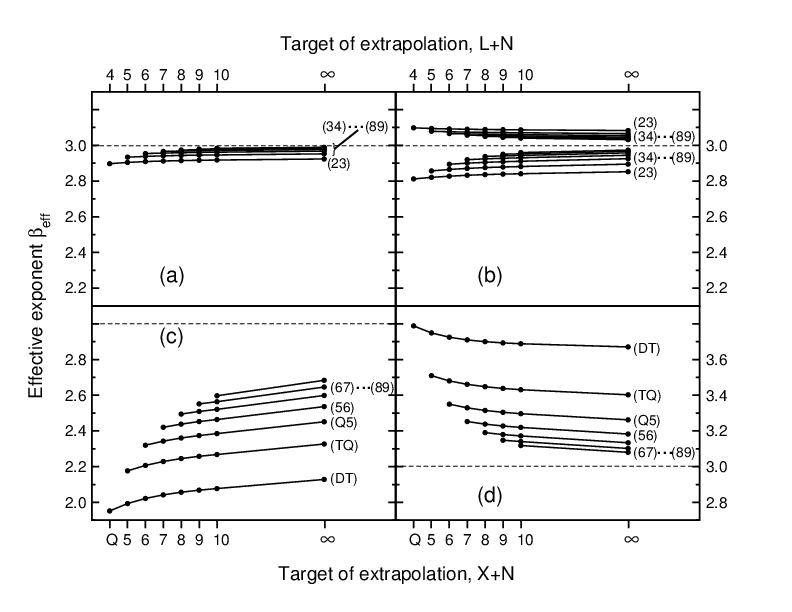
The electron correlation energy of two-electron atoms is known to
converge asymptotically as (L+1) -3 to the complete basis set limit,
where L is the maximum angular momentum quantum number included in the
basis set. Numerical evidence has established a similar asymptotic
convergence X -3 with the cardinal number X of correlation-consistent
basis sets cc-pVXZ for coupled cluster singles and doubles (CCSD) and
second order perturbation theory (MP2) calculations of molecules. The
main focus of this article is to probe for deviations from asymptotic
convergence behavior for practical values of X by defining a trial
function X - β that for an effective exponent β=βeff(X,X+1,X+N)
provides the correct energy EX+N, when extrapolating from results for
two smaller basis sets, EX and E{X+1}. This analysis is first applied
to "model" expansions available from analytical theory, and then to a
large body of finite basis set results (X=D,T,Q,5,6) for 105 molecules
containing H, C, N, O, and F, complemented by a smaller set of 14 molecules
for which accurate complete basis set limits are available from MP2-R12
and CCSD-R12 calculations. βeff is generally found to vary monotonically
with the target of extrapolation, X+N, making results for large but finite
basis sets a useful addition to the limited number of cases where complete
basis set limits are available. Significant differences in effective
convergence behavior are observed between MP2 and CCSD (valence) correlation
energies, between hydrogen-rich and hydrogen-free molecules, and, for He,
between partial-wave expansions and correlation-consistent basis sets.
Deviations from asymptotic convergence behavior tend to get smaller as X
increases, but not always monotonically, and are still quite noticeable
even for X=5. Finally, correlation contributions to atomization energies
(rather than total energies) exhibit a much larger variation of effective
convergence behavior, and extrapolations from small basis sets are found
to be particularly erratic for molecules containing several electronegative
atoms. Observed effects are discussed in the light of results known from
analytical theory. A carefully calibrated protocol for extrapolations to
the complete basis set limit is presented, based on a single "optimal"
exponent βopt(X,X+1,∞) for the entire set of molecules, and
compared to similar approaches reported in the literature.
|
|
van Gunsteren, W. F.; Bakowies, D.; Baron, R. et al.,
"
Biomolecular modeling: Goals, problems, perspectives
"
Angew. Chem. Int. Ed. 2006, 45, 4064-4092, Angew. Chem. 2006, 118, 4168-4198.
Citing Articles (ISI)
Journal Page
Abstracts off

Computation based on molecular models is playing an increasingly
important role in biology, biological chemistry, and biophysics. Since
only a very limited number of properties of biomolecular systems is
actually accessible to measurement by experimental means, computer
simulation can complement experiment by providing not only averages,
but also distributions and time series of any definable quantity,
for example, conformational distributions or interactions between
parts of systems. Present day biomolecular modeling is limited in
its application by four main problems: 1) the force-field problem,
2) the search (sampling) problem, 3) the ensemble (sampling) problem,
and 4) the experimental problem. These four problems are discussed and
illustrated by practical examples. Perspectives are also outlined for
pushing forward the limitations of biomolecular modeling.
|
|
Bakowies, D. ,
"
Atomization energies from ab initio calculations without empirical corrections
"
Scientific Report of the Swiss National Supercomputing Centre 2005, 14-17.
Scientific Report
Abstracts off
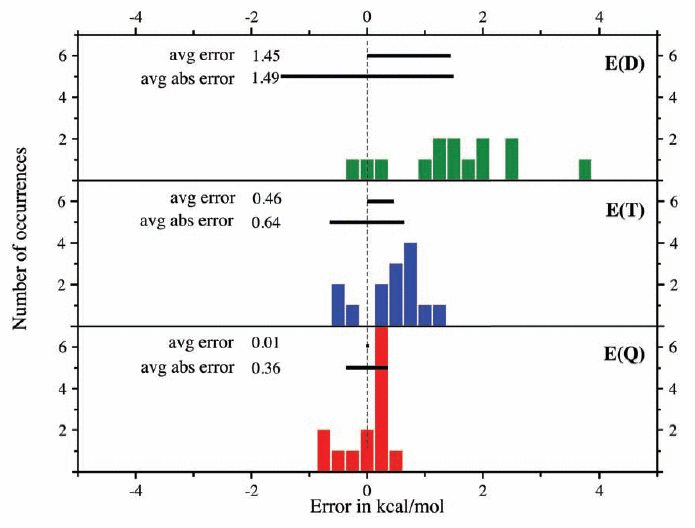
Accurate thermochemistry has turned out to be a
major challenge for standard ab initio quantum
chemistry. High levels of electron correlation combined
with very large basis sets are required to
adequately treat the formation of a molecule from
its constituent atoms. The application of current
approaches is thus limited to very small molecules
unless empirical corrections are permitted to account
for the average effects of basis set incompleteness
and higher order electron correlation.
Here we test economical compound methods
based entirely on ab initio electronic structure theory
and void of empirical corrections, taking advantage
of the observations that (a) higher-order
electron correlation corrections are much less basis-
set dependent than low-order (MP2) correlation
energies, and (b) basis set incompleteness
errors can largely be eliminated through extrapolation
techniques. These compound methods should
be accurate enough even for larger molecules so
that they provide useful references for the parametrization
of more approximate methods, particularly
in semiempirical quantum chemistry.
|
|
Christen, M.; Hünenberger, P. H.; Bakowies, D. et al.,
"
The GROMOS software for biomolecular simulation: GROMOS05
"
J. Comput. Chem. 2005, 26, 1719-1751.
Citing Articles (ISI)
Journal Page
Abstracts off
We present the latest version of the Groningen Molecular Simulation
program package, GROMOS05. It has been developed for the dynamical
modelling of (bio)molecules using the methods of molecular dynamics,
stochastic dynamics, and energy minimization. An overview of GROMOS05
is given, highlighting features not present in the last major release,
GROMOS96. The organization of the program package is outlined and the
included analysis package GROMOS++ is described. Finally, some applications
illustrating the various available functionalities are presented.
|
|
Chandrasekhar, I.; Bakowies, D.; Glättli, A.; Hünenberger, P.; Pereira, C.; van Gunsteren, W. F.,
"
Molecular dynamics simulation of lipid bilayers with GROMOS96: Application of surface tension
"
Mol. Simul. 2005, 31, 543-548.
Citing Articles (ISI)
Journal Page
Abstracts off
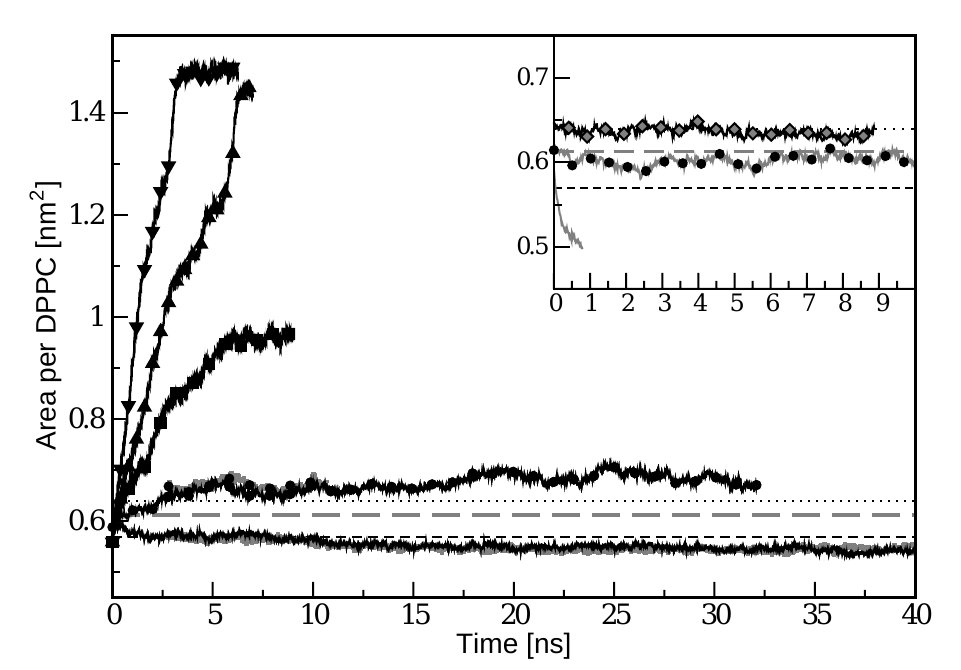
The GROMOS96 force fields 45A3 and 53A5, when applied to
dipalmitoylphosphatidylcholine (DPPC) membranes, have a tendency
to result in a reduced area per lipid in constant pressure
simulations. The application of surface tension is effective in
increasing the area per lipid, a measure of the phase of the
membrane, but only if the area is already close to the experimental
range. Therefore the surface tension cannot compensate for strong
inadequacies in the force-field parameters. The behaviour of the 45A3
force field from long NPnγT simulations of tens of nanoseconds is
analysed over a range of different surface tensions. Comparisons
are made with the corresponding NPnAT simulations.
|
|
Baron, R.; Bakowies, D.; van Gunsteren, W. F.,
"
Principles of carbopeptoid folding: A molecular dynamics simulation study
"
J. Peptide Sci. 2005, 11, 74-84.
Citing Articles (ISI)
Journal Page
Abstracts off
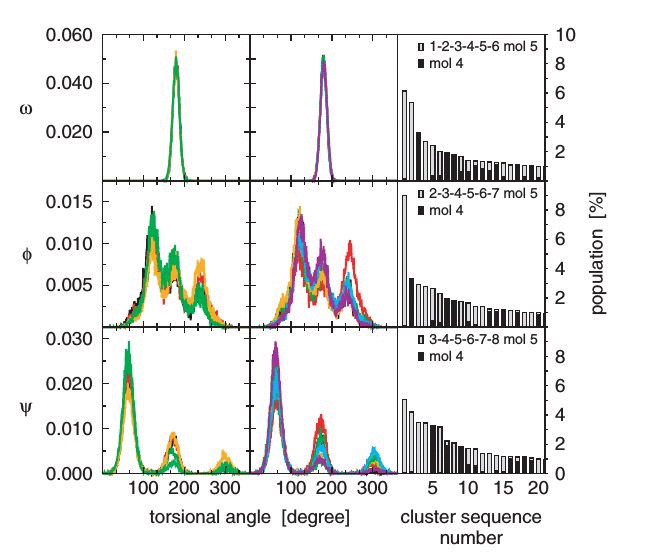
The conformational spaces of five oligomers of tetrahydrofuran-based
carbopeptoids in chloroform and dimethyl sulfoxide were investigated
through nine molecular dynamics simulations. Prompted by nuclear
magnetic resonance experiments that indicated various stable folds
for some but not all of these carbopeptoids, their folding behaviour
was investigated as a function of stereochemistry, chain length and
solvent. The conformational distributions of these molecules were
analysed in terms of occurrence of hydrogen bonds, backbone torsional-angle
distributions, conformational clustering and solute configurational
entropy. While a cis-linkage across the tetrahydrofuran ring favours
right-handed helical structures, a trans-linkage results in a larger
conformational variability. Intra-solute hydrogen bonding is reduced
with increasing chain length and with increasing solvent polarity.
Solute configurational entropies confirm the picture obtained: they
are smaller for cis- than for trans-linked peptides, for chloroform
than for dimethyl sulfoxide as solvent and for shorter peptide chains.
The simulations provide an atomic picture of molecular conformational
variability that is consistent with the available experimental data.
|
|
Baron, R.; Bakowies, D.; van Gunsteren, W. F.,
"
Carbopeptoid folding: Effects of stereochemistry, chain length, and solvent
"
Angew. Chem. Int. Ed. 2004, 43, 4055-4059, Angew. Chem. 2004, 116, 4147-4151.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
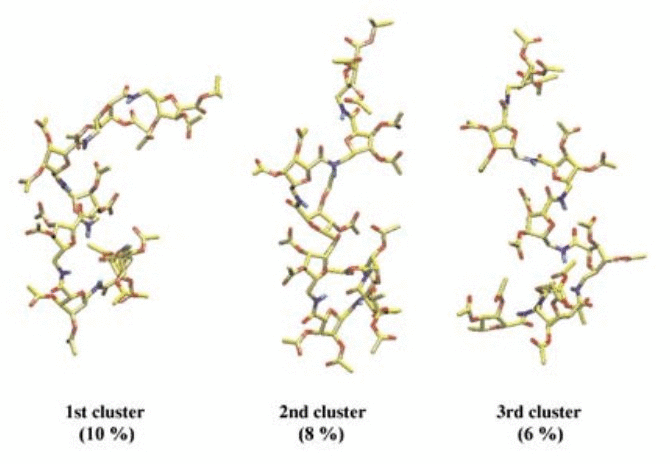
No abstract.
|
|
Daura, X.; Bakowies, D.; Seebach, D.; Fleischhauer, J.; van Gunsteren, W. F.; Krüger, P.,
"
Circular dichroism spectra of β-peptides: Sensitivity to molecular structure and effects of motional averaging
"
Eur. Biophys. J. 2003, 32, 661-670.
Citing Articles (ISI)
Journal Page
Abstracts off
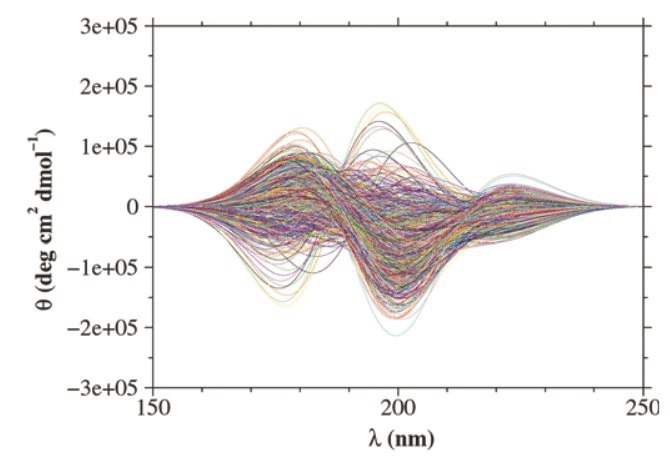
Circular dichroism spectra of two β-peptides, i.e. peptides composed
of β-amino acids, calculated using ensembles of configurations obtained
by molecular dynamics simulation are presented. The calculations were
based on 200 ns simulations of a β-heptapeptide in methanol at 298 K
and 340 K and a 50 ns simulation of a β-hexapeptide in methanol at
340 K. In the simulations the peptides sampled both folded (helical)
and unfolded structures. Trajectory structures with common backbone
conformations were identified and grouped into clusters. The CD spectra
were calculated for individual structures, based on peptide-group dipole
transition moments obtained from semi-empirical molecular orbital theory
and using the so-called matrix method. The single-structure spectra were
then averaged over entire trajectories and over clusters of structures.
Although certain features of the experimental CD spectra of the
β-peptides are reproduced by the trajectory-average spectra, there
exist clear differences between the two sets of spectra in both wavelength
and peak intensities. The analysis of individual contributions to the
average spectra shows that, in general, the interpretation of a CD signal
in terms of a single structure is not possible. Moreover, there is a large
variation in the CD spectra calculated for a set of individual structures
that belong to the same cluster, even when a structurally tight clustering
criterion is used. This indicates that the CD spectra of these peptides
are very sensitive to small local structural differences.
|
|
Bakowies, D.
"
Biomolekulare Reality-Simulationen
"
Nachr. Chem. 2003, 51, 788-793.
Citing Articles (ISI)
Journal Page
Abstracts off

"Observe while it happens": Während man früher häufig auf vereinfachte
Kraftfelddarstellungen zurückgriff und Energiebarrieren künstlich durch
Anlegen von Zwangskräften überwand, sehen wir dank schnellerer Rechner
heute schon die ersten Simulationen der Membranaggregation, des
Wassertransports durch Membrankanäle und der Peptid- und Proteinfaltung
im vollen atomaren Detail.
"Observe while it happens": While we have been used to simplified force
field descriptions and artificial bias forces enabling us to surmount
energy barriers, we are now seeing - thanks to faster computers - the
first simulations providing full atomistic detail of membrane aggregation,
of water transport through membrane channels and of peptide and protein
folding.
|
|
Bakowies, D.
"
Trendbericht Theoretische Chemie 2002: Kraftfelder für biomolekulare Simulationen
"
Nachr. Chem. 2003, 51, 325-327.
Citing Articles (ISI)
Journal Page
Abstracts off

No abstract.
|
|
Bakowies, D.
"
Analyzing solvent in protein cavities: Methods and application to fatty acid binding protein
"
Annual Report of the Competence Center for Computational Chemistry, ETH Zürich 2003, 24-43.
Journal Page
Abstracts off
(Technical Report on a Feature Project)
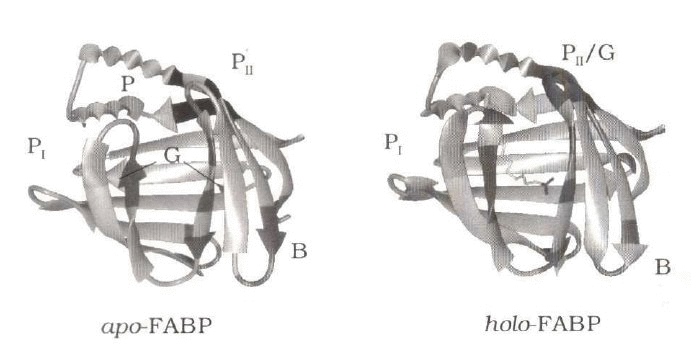
In this paper we present a method to generate a closed surface
which embraces a set of predefined atoms. The central idea is to
construct a polyhedron whose vertices represent the set of chosen
atoms and to find all solvent molecules enclosed by the polyhedron.
The proposed procedure entails five steps: (1) Choice of a set of
protein atoms which define the boundaries of the protein interior,
the protein cavity, or more generally the protein region of interest.
(2) Projection of this set of atoms onto a sphere around their
geometrical center. (3) Triangulation of the spherical surface.
(4) Back transformation to the original coordinate system.
(5) Identification of solvent molecules inside the so-generated
polyhedron.
This procedure is applied to 5 ns long MD trajectories of apo- and
holo-fatty acid binding protein (FABP). The beta-barrel type structure
of FABP encompasses a large solvent-filled cavity, and only some of
the internal water is expelled upon introduction of the palmitate
ligand. In this report we concentrate on structural and dynamical
aspects of internal water and on differences between the apo and holo
forms of the protein.
|
|
Baron, R.; Bakowies, D.; van Gunsteren, W. F.; Daura, X.,
"
β-peptides with different secondary-structure preferences: How different are their conformational spaces?
"
Helv. Chim. Acta 2002, 85, 3872-3882.
Citing Articles (ISI)
Journal Page
Abstracts off
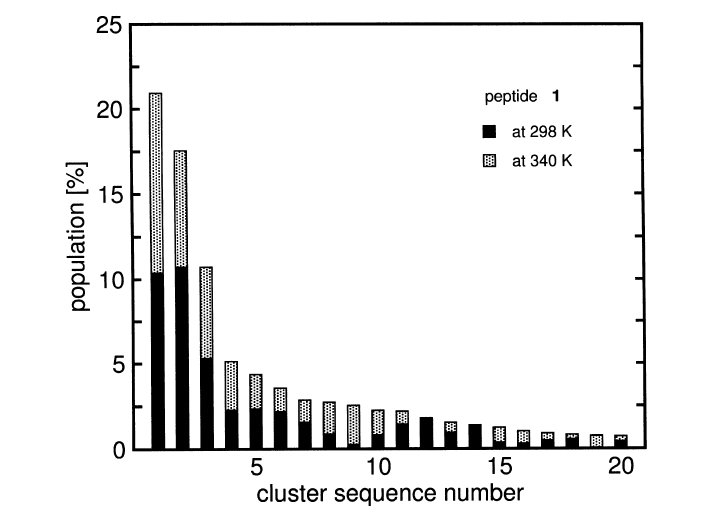
The conformational spaces accessible to two beta-hexapeptides in MeOH
at 298 K and 340 K are investigated by molecular-dynamics simulation
with an atomistic model of both solute and solvent. The structural
properties of these peptides have been previously studied by NMR
in MeOH at room temperature. The experimental data could be fitted
to a model (P)-12/10-helix for one of the peptides and a model hairpin
with a ten-membered H-bonded turn for the other. The goal of the present
work is to determine whether the conformational spaces accessible to
these two peptides of seemingly different conformational properties
contain any common regions. In other words, to what extent are the
evident differences found at the macroscopic level also present at
the microscopic structural level? It is found that, for the two
peptides studied, the conformational spaces sampled in the respective
simulations show significant overlap.
|
|
Bakowies, D.; van Gunsteren, W. F.,
"
Water in protein cavities: A procedure to identify internal water
and exchange pathways and application to fatty acid binding protein
"
Proteins 2002, 47, 534-545.
Citing Articles (ISI)
Journal Page
Abstracts off
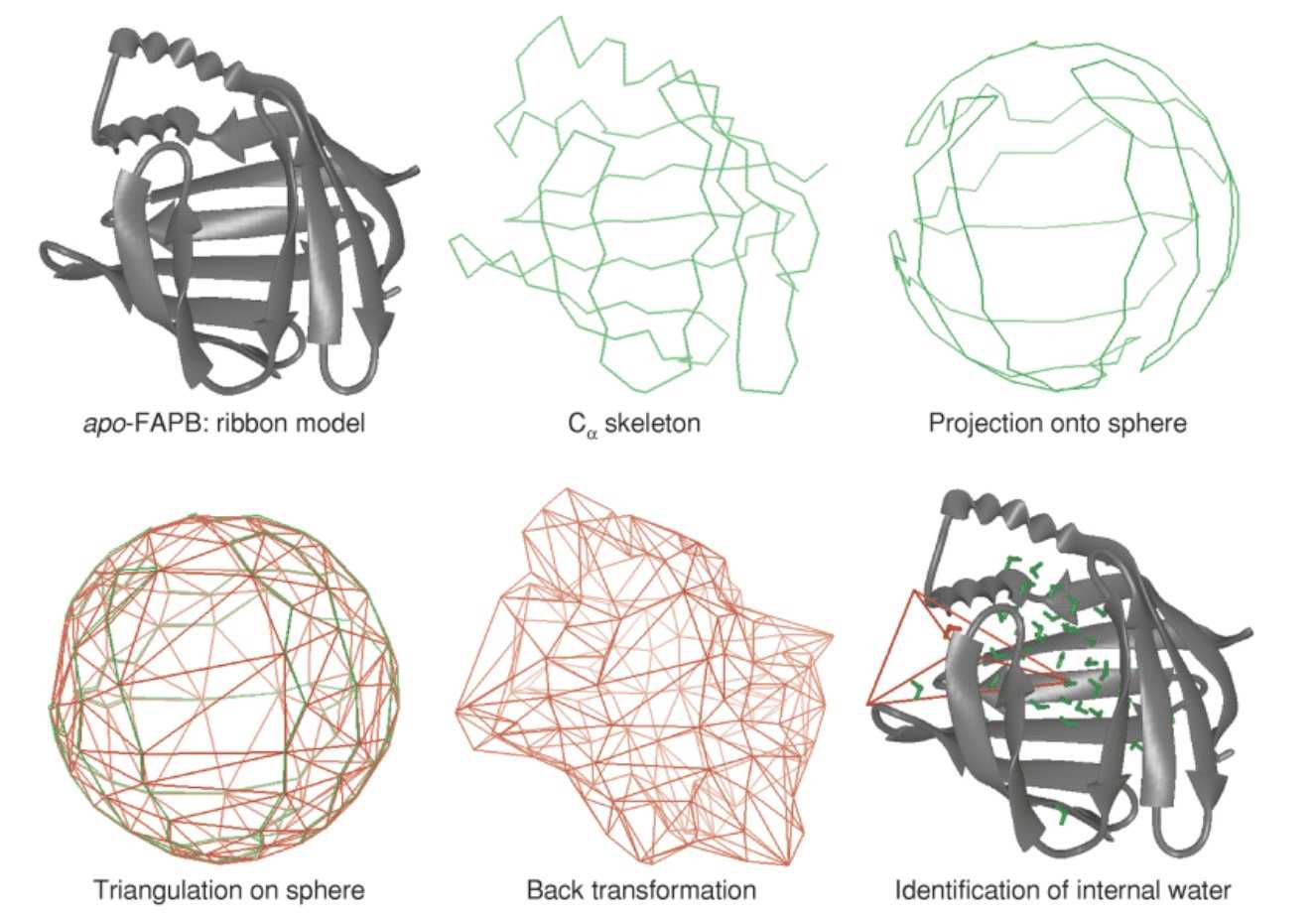
A computational approach based on Delaunay triangulation is presented
to identify internal water molecules in proteins and to capture pathways
of exchange with the bulk. The implemented procedure is computationally
efficient and can easily be applied to long molecular dynamics trajectories
of protein simulations. In an application to fatty acid-binding protein in
apo-form and with bound palmitate, several protein orifices known from
crystal structures have been confirmed to be major portals of solvent
exchange. Differences between the two forms of the protein are observed
and discussed.
|
|
Bakowies, D.; van Gunsteren, W. F.,
"
Simulations of apo and holo-fatty acid binding protein:
Structure and dynamics of protein, ligand and internal water
"
J. Mol. Biol. 2002, 315, 713-736.
Citing Articles (ISI)
Journal Page
Abstracts off
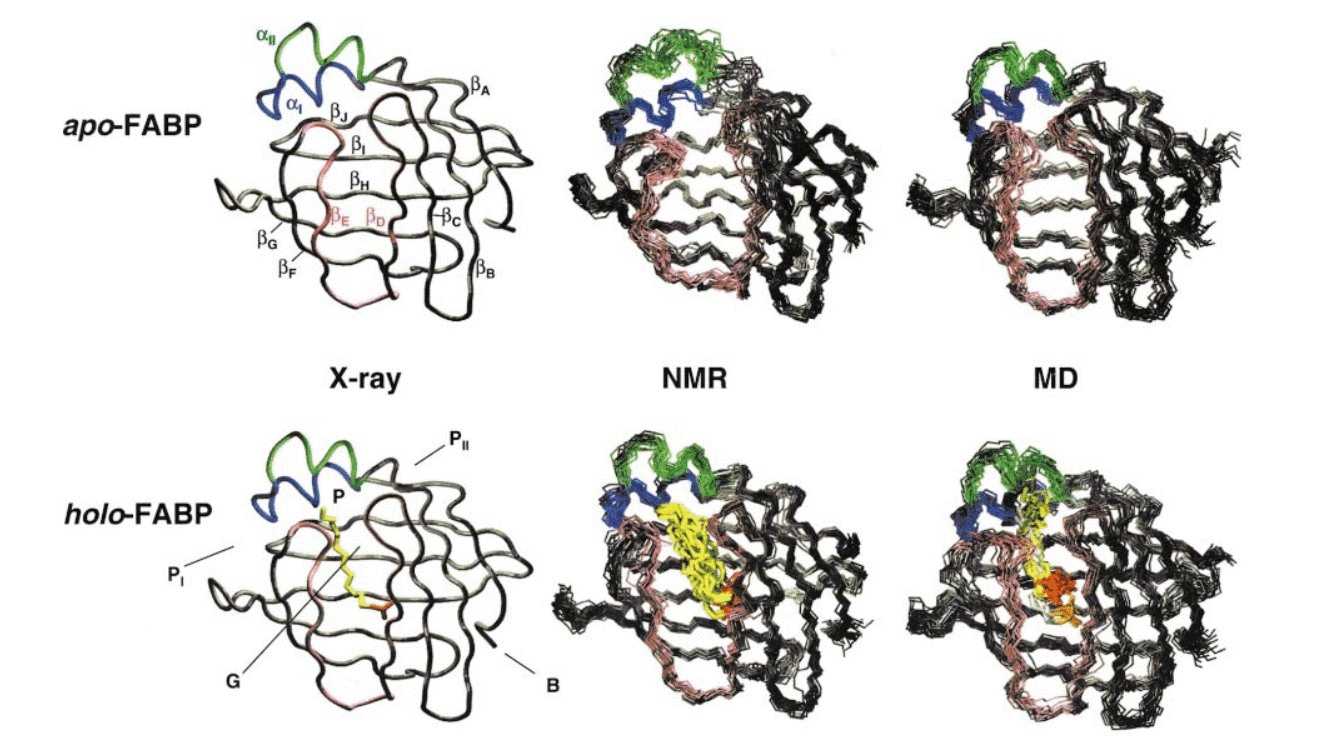
Two molecular dynamics simulations of 5 ns each have been carried out for
rat intestinal fatty acid binding protein, in apo-form and with bound
palmitate. The fatty acid and a number of water molecules are encapsulated
in a large interior cavity of the barrel-shaped protein. The simulations
are compared to experimental data and analyzed in terms of root mean square
deviations, atomic β-factors, secondary structure elements, hydrogen bond
patterns, and distance constraints derived from nuclear Overhauser experiments.
Excellent agreement is found between simulated and experimental solution
structures of holo-FABP, but a number of differences are observed for the
apo-form. The ligand in holo-FABP shows considerable displacement after
about 1.5 ns and displays significant configurational entropy. A novel
computational approach has been employed to identify internal water and
to capture exchange pathways. Orifices in the portal and gap regions of
the protein, discussed in the experimental literature, have been confirmed
as major openings for solvent exchange between the internal cavity and bulk
water. A third opening on the opposite side of the barrel experiences
significant exchange but it does not provide a pathway for further passage
to the central cavity. Internal water is characterized in terms of density
distributions, interaction energies, mobility, protein contact times, and
water molecule coordination. A number of differences are observed between
the apo and holo-forms and related to differences in the protein structure.
Solvent inside apo-FABP, for example, shows characteristics of a water
droplet, while solvent in holo-FABP benefits from interactions with the
ligand headgroup and slightly stronger interactions with protein residues.
|
|
Gunsteren, W. F.; Bakowies, D.; Bürgi, R. et al.,
"
Molecular dynamics simulation of biomolecular systems
"
Chimia 2001, 55, 856-860.
Citing Articles (ISI)
Journal Page
Abstracts off
The group for computer-aided chemistry at the ETH Zurich focuses its
research on the development of methodology to simulate the behavior
of biomolecular systems and the use of simulation techniques to
analyze and understand biomolecular processes at the atomic level.
Here, the current research directions are briefly reviewed and
illustrated with a few examples.
|
|
van Gunsteren, W. F.; Bakowies, D.; Damm, W.; Hansson, T; Stocker, U.; Daura, X.,
"
Practical aspects of simulation studies of biomolecular systems
"
in Dynamics, structure and function of biological macromolecules,
edited by Jardetzky, O. and Finucane, M., NATO ASI Series A315, IOS Press, Amsterdam, 2001, pp. 1-26.
Citing Articles (ISI)
Abstracts off
With the ever-continuing increase of the power of computers, simulation
of biomolecular systems in atomic detail has come of age. It is nowadays
possible to simulate the classical dynamics of a protein solvated in
aqueous solution over time periods of several nanoseconds. The classical
equations of motion for the 104-105 atoms in the system can be integrated
forward in time for millions of time steps of the order of 1 fs. This
allows the study of the conformational equilibrium and dynamics of a
variety of biomolecular systems, such as membranes, micelles, protein or
DNA complexes. Once the reliability of the molecular models, force fields
and computational procedures has been established by comparison of simulated
properties with known experimental ones, molecular dynamics (MD) simulation
can be a very powerful method to predict molecular properties that are
inaccessible to experimental probes. It provides a microscopic picture of,
in principle, unlimited resolution in time, space and energy. Secondly, it
allows for the study of systems or processes that are impossible to create
in reality. System parameters can be changed at will to study particular
cause-effect relationships, leading to an enhanced understanding of
biomolecular systems, which are generally of complex nature. A biomolecular
simulation study possesses a number of practical aspects, which require
choices of parameters, procedures and approximations to be made.
1. Molecular model, choice of spatial degrees of freedom, spatial boundary
conditions, system sizes and equations of motion.
2. Interaction between the particles or atoms, or along the degrees of
freedom.
3. Simulation set-up, treatment of long-range forces, coupling to
temperature or pressure baths, choice of initial structure and time step.
4. Simulation software, its reliability, efficiency and capabilities.
5. Convergence, statistics and sampling of molecular or system properties.
6. Comparison of simulated properties with experimental data,
(in)sensitivity of properties to molecular conformation.
7. Analysis and interpretation of simulated trajectories or ensembles.
In this paper, these seven aspects of biomolecular simulation are briefly
discussed and illustrated with examples taken from the literature.
|
|
Kollman, P. A.; Kuhn, B.; Donini, O.; Peräkylä, M.; Stanton, R.; Bakowies, D.,
"
Elucidating the nature of enzyme catalysis utilizing a new twist on an old methodology: Quantum
mechanical - free energy calculations on chemical reactions in enzymes and in aqueous solution
"
Acc. Chem. Res. 2001, 34, 72-79.
Citing Articles (ISI)
Journal Page
Abstracts off
How do enzymes achieve very large rate enhancements compared to
corresponding uncatalyzed reactions in solution? We present a
computational approach which combines high-level ab initio quantum
mechanical calculations with classical free energy calculations to
address this question. Our calculations lead to accurate estimates
of ΔG&Dagger for both trypsin and catechol O-methyltransferase-catalyzed
and reference uncatalyzed reactions and give new insights into the
nature of enzyme catalysis. The same methodology applied to steps
in the catalytic mechanism of citrate synthase further supports
the conclusion that one need not invoke special concepts such as
"low-barrier hydrogen bonds" or "pKa matching" to explain enzyme
catalysis.
|
|
Bakowies, D.; Kollman, P. A.,
"
Theoretical study of base-catalyzed amide hydrolysis:
Gas and aqueous phase hydrolysis of formamide
"
J. Am. Chem. Soc. 1999, 121, 5712-5726.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
Base-catalyzed hydrolysis of formamide in the gas phase and in aqueous
solution has been studied using a combination of quantum chemical and
statistical mechanical methods. A three-step procedure has been applied
which comprises the determination of a gas-phase reaction path by high-level
ab initio calculations, the calibration of empirical soluteâsolvent
potentials, and classical Monte Carlo simulations of the solute immersed
in a bath of solvent molecules. These simulations yield the solvent effect
as a potential of mean force along the predetermined reaction coordinate.
Each of the three consecutive steps of base-catalyzed hydrolysis has been
analyzed in detail: the formation of a tetrahedral intermediate, its
conformational isomerization, and the subsequent breakdown to products. The
reaction is very exothermic in the gas phase and involves only moderate
barriers for the latter two steps. Aqueous solvent, however, induces a
significant barrier toward formation of the intermediate. On the other hand,
it also facilitates conformational isomerization and produces a more
product-like transition state for the breakdown step. Solvent effects,
as expressed by differences in free energy of solvation, are found to
reflect variations in the solute's charge distribution and are readily
explained by the analysis of hydrogen bond patterns. The calculated
free energy profile is in satisfactory agreement with available
experimental data for the solution-phase reaction.
|
|
Stanton, R. V.; Peräkylä, M.; Bakowies, D.; Kollman, P. A.,
"
Combined ab initio and free energy calculations to study reactions in enzymes
and solution: Amide hydrolysis in trypsin and aqueous solution
"
J. Am. Chem. Soc. 1998, 120, 3448-3457.
Citing Articles (ISI)
Journal Page
Abstracts off
We present a new more general way to combine ab initio quantum mechanical
calculations with classical mechanical free energy perturbation approach
to calculate the energetics of enzyme-catalyzed reactions and the same
reaction in solution. This approach, which enables enzyme and solution
reactions to be compared without the use of empirical parameters, is
applied to the formation of the tetrahedral intermediate in trypsin,
but it should be generally applicable to any enzymatic reaction. Critical
to the accurate calculation of the reaction energetics in solution is the
estimate of the free energy to assemble the reacting groups, where the
approach recently published by Hermans and Wang (J. Am. Chem. Soc. 1997,
119, 2707) was used. A central aspect of this new approach is the use of
the RESP protocol to calculate the charge distribution of structures along
the reaction pathway, which enables us to circumvent problems in partitioning
the charge across a residue that is being divided into QM and MM parts. The
classical mechanical free energy calculations are implemented with two
different approaches, "Cartesian mapping" and "flexible FEP". The similarity
of the results found by using these two approaches supports the robustness
of the calculated free energies. The calculated free energies are in quite
good agreement with available experimental data for the activation free
energies in the enzyme and aqueous phase reactions.
|
|
Bakowies, D.; Thiel, W.,
"
Hybrid models for combined quantum mechanical and molecular mechanical approaches
"
J. Phys. Chem. 1996, 100, 10580-10594.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
A hierarchy of three models for combined quantum mechanical (QM) and
molecular mechanical (MM) approaches is presented. They simplify the
QM description of large molecules by reducing it to the electronically
important fragment which interacts with the molecular mechanically
treated remainder of the molecule. In the simplest model A, the QM
fragments are only mechanically embedded in their MM environment. The
more refined models B and C include a quantum mechanical treatment of
electrostatic interactions between the fragments and a semiclassical
description of polarization. The implementation of models A-C for MNDO
type wavefunctions and the MM3 force field is outlined. Selected
applications in organic chemistry are discussed, addressing the ability
of the proposed models to reproduce substituent effects (MM) on chemical
structure and reactivity (QM). These applications include protonations,
deprotonations, hydride transfer reactions, nucleophilic additions, and
nucleophilic ring cleavage reactions.
|
|
Bakowies, D.; Thiel, W.,
"
Semiempirical treatment of electrostatic potentials and partial charges in
combined quantum mechanical and molecular mechanical approaches
"
J. Comput. Chem. 1996, 17, 87-108.
Citing Articles (ISI)
Journal Page
Abstracts off
A semiempirical treatment of electrostatic potentials and partial charges
is presented. These are the basic components needed for the evaluation of
electrostatic interaction energies in combined quantum mechanical and
molecular mechanical approaches. The procedure to compute electrostatic
potentials uses AM1 and MNDO wave functions and is based on one previously
suggested by Ford and Wang. It retains the NDDO approximation and is thus
both easy to implement and computationally efficient. Partial atomic charges
are derived from a semiempirical charge equilibration model, which is based
on the principle of electronegativity equalization. Large sets of ab initio
restricted Hartee-Fock (RHF/6-31G*) reference data have been used to
calibrate the semiempirical models. Applying the final parameters
(C, H, N, O), the ab initio electrostatic potentials are reproduced
with an average accuracy of 20% (AM1) and 25% (MNDO), respectively, and
the ab initio potential derived charges normally to within 0.1 e. In most
cases our parameterized models are more accurate than the much more
expensive quasi ab initio techniques, which employ deorthogonalized
semiempirical wave functions and have generally been preferred in
previous applications.
|
|
Bakowies, D.; Bühl, M.; Patchkovskii, S.; Thiel, W.,
"
Theoretical studies on giant fullerenes and on endohedral fullerene complexes
"
in Fullerenes: Recent advances in the physics and chemistry of fullerenes and
related materials, Vol. 3, edited by Ruoff, R. S. and Kadish, K. M., The
Electrochemical Society, Pennington, NJ, 1996, pp. 901-910.
Abstracts off
Ab initio SCF, density functional, and semiempirical methods have been
used to study selected topics from fullerene chemistry. These include
the structural preferences and the stabilities of large icosahedral
fullerenes (C180, C240, C540, C960), the mechanism of incorporating helium
into C60 to form the endohedral complex He@C60, and the NMR chemical shifts
of 3He in endohedral complexes involving fullerenes of different sizes as
well as C60H36. Our computational results are discussed in relation to
other theoretical and experimental work.
|
|
Bakowies, D.; Bühl, M.; Thiel, W.,
"
A density functional study on the shape of C180 and C240 fullerenes
"
Chem. Phys. Lett. 1995, 247, 491-493.
Citing Articles (ISI)
Journal Page
Abstracts off
At the gradient-corrected BP86/SV level of density functional theory,
the fully optimised, facetted geometry of Ih-C180 is 126 kcal/mol
lower in energy than an optimised spherical structure where all atoms
are constrained to lie on the same sphere. Likewise, using MNDO geometries,
facetted Ih-C240 is more stable than the constrained spherical form by 203
and 202 kcal/mol at the non-local BP86/SV and the local VWN/SV levels,
respectively. These findings are at variance with predictions from local
density functional calculations employing the divide-and-conquer
approximation and the Harris functional, but confirm the results of
recent MNDO and ab initio SCF studies.
|
|
Bakowies, D.; Bühl, M.; Thiel, W.,
"
Can large fullerenes be spherical ?
"
J. Am. Chem. Soc. 1995, 117, 10113-10118.
Citing Articles (ISI)
Journal Page
Abstracts off
MNDO geometry optimizations predict a single energy minimum for each
of the Goldberg type (Ih) fullerenes C180, C240, C540, and C960 which
corresponds to an icosahedrally shaped structure with strong curvature
at the 12 pentagons and nearly planar segments composed of hexagons.
Constrained geometry optimizations preserving a spherical shape lead
to considerably larger energies and show that an evenly distributed
curvature is strongly disfavored. The results are confirmed quantitatively
by ab initio SCF calculations for C180 and C240 employing a split valence
basis set, but contrast the conclusions from a previous density functional
study. The observed trends are discussed on the basis of curvature-corrected
Hückel calculations and simple force field estimates.
|
|
Slanina, Z.; François, J.-P.; Kolb, M.; Bakowies, D.; Thiel, W.,
"
Calculated relative stabilities of C84
"
Fullerene Sci. Techn. 1993, 1, 221-230.
Journal Page
Abstracts off
C84 is treated as a system composed of 24 local minima whose energies,
geometries, and vibrational frequencies are obtained from MNDO
calculations. The predicted global minimum of D2d symmetry remains the
most abundant species in the equilibrium isomeric mixture only till
276 K, being replaced by a D2 species beyond that point. However, a C1
structure is prevalent in the high temperature limit. The calculated
composition around a temperature of 1000 K is consistent with very recent
NMR observations.
|
|
Slanina, Z.; François, J.-P.; Bakowies, D.; Thiel, W.,
"
Fullerene C78 isomers: Temperature dependence of their calculated relative stabilities
"
J. Mol. Struct.: THEOCHEM 1993, 279, 213-216.
Citing Articles (ISI)
Journal Page
Abstracts off
C78 is treated as a system composed of five local minima (D3h(I), D3h(II),
D3, D2v(I) and C2v(II)) whose energies, geometries and vibrational
frequencies are obtained from MNDO calculations. The AM1 and PM3 energy
data also considered. The predicted global minimum (C2v(II)) remains the
most stable species of the equilibrium isomeric mixture even at high
temperatures, and the D3n (I) structure is negligible throughout. The
remaining three species exhibit comparable stabilities and make up nearly
50% of the high temperature mixture. The recently observed ratio of 1:5
of the D3 and C2v (I) structures is reached at temperatures of 1308 K,
734 K and 980 K for the MNDO, AM1 and PM3 energetics respectively.
|
|
Bakowies, D.; Kolb, M.; Thiel, W.; Richard, S.; Ahlrichs, R.; Kappes, M. M.,
"
Quantum chemical study of C84 fullerene isomers
"
Chem. Phys. Lett. 1992, 200, 411-417.
Citing Articles (ISI)
Journal Page
Abstracts off
Semiempirical and ab initio SCF calculations are reported for the
C84 fullerenes with isolated pentagons. The optimized geometries
and relative stabilities are discussed. All methods applied predict
two nearly isoenergetic structures with D2 and D2d symmetry to be the
most stable of the 24 isomers considered, which is consistent with the
experimental observed 13C-NMR spectrum. Infrared spectra are predicted
for these D2 and D2d isomers. The semiempirical results (MNDO, AM1, PM3)
for the geometries and relative energies are in excellent agreement with
the ab initio predictions at the split-valence SCF level.
|
|
Bakowies, D.; Gelessus, A.; Thiel, W.,
"
Quantum chemical study of C78 fullerene isomers
"
Chem. Phys. Lett. 1992, 197, 324-329.
Citing Articles (ISI)
Journal Page
Abstracts off
Semi-empirical and ab initio calculations are reported for the five
C78 fullerenes with isolated pentagons. The optimized geometries
and relative stabilities are discussed. The D3h structure previously
favored on the basis of simple Hückel arguments is found to be the
least stable isomer at all theoretical levels applied. The most stable
isomer corresponds to a C2v structure which has recently been observed
experimentally together with two other isomers. Infrared spectra are
predicted for all five isomers.
|
|
Slanina, Z.; Adamowicz, L.; Bakowies, D.; Thiel, W.,
"
Fullerene C50 isomers: Temperature-induced interchange of relative stabilities
"
Thermochim. Acta 1992, 202, 249-254.
Citing Articles (ISI)
Journal Page
Abstracts off
The species C50 is treated as a system composed of three local energy
minima (D5h, D3, and C2v) found in recent modified neglect of diatomic
overlap (MNDO) calculations. Although the D5h species corresponds to
the deepest minimum it is the most stable structure only up to about
1390 K. Beyond this temperature the D3 species becomes relatively
more populous.
|
|
Bakowies, D.; Thiel, W.,
"
Theoretical study of Buckminsterfullerene derivatives C60Xn (X=H, F; n = 2, 36, 60)
"
Chem. Phys. Lett. 1992, 192, 236-242.
Citing Articles (ISI)
Journal Page
Abstracts off
Semi-empirical SCF calculations at the MNDO, AM1, and PM3 levels are
reported for the title compounds. The predicted relative stabilities
are discussed for all 18 clusters studied. The calculated equilibrium
geometries and vibrational spectra are presented for C60X36(Th) and
C60X60(Ih). Contrary to a previous suggestion, C60H60 and C60F60 prefer
an icosahedral Ih structure over a distorted I structure. The calculated
bond dissociation enthalpies, equilibrium bond lengths, and vibrational
frequencies indicate a reduced C---X bond strength in C60X60(Ih).
|
|
Bakowies, D.; Thiel, W.,
"
MNDO study of large carbon clusters
"
J. Am. Chem. Soc. 1991, 113, 3704-3714.
Citing Articles (ISI)
Journal Page
Supplementary Material
Abstracts off
MNDO calculations with complete geometry optimization are reported for
30 polyhedral carbon clusters Cn (20 ≤ n ≤ 540). The MNDO results for
a planar graphite sheet are extrapolated from calculations on D6h
hydrocarbons CnHm (n=6k2, m=6k, k=1...6) and used as reference for
discussing the properties of the clusters. The structural features of the
clusters are correlated with their stability. The relative MNDO energies
with respect to graphite are compared with curvature-corrected Hückel
calculations and with force field estimates, and criteria for the stability
of the clusters are discussed. Infrared spectra are predicted for six stable
clusters. Several cationic lithium complexes and their interconversions are
investigated for C60 and C42. Finally, computational aspects and performance
data are considered, particularly for the largest clusters studied.
|
|
Bakowies, D.; Thiel, W.,
"
Theoretical infrared spectra of large carbon clusters
"
Chem. Phys. 1991, 151, 309-321.
Citing Articles (ISI)
Journal Page
Abstracts off
Harmonic force constant calculations at the MNDO SCF level are
reported for 22 polyhedral carbon clusters Cn (20 ≤ n ≤ 240) and
for the reference molecules benzene, coronene, and corannulene. New
or revised assignments are suggested for coronene and corannulene.
Based on the results for the reference molecules and for C60, MNDO
is expected to overestimate the vibrational frequencies of the unknown
clusters by 10% and to yield reasonable intensity patterns. Calculated
infrared spectra are shown and discussed for 12 carbon clusters, with
particular emphasis on those which might be observable spectroscopically.
The zero-point vibrational energies and the entropy contributions to the
free enthalpy are only of minor importance for the thermodynamic
stabilities of different clusters.
|
|
Requirements to follow links on this page
Article download:
Electronic journal subscription
Citing Articles (ISI):
Subscription to ISI Web of Knowledge, open ISI session
|
|